Topic Menu
► Topic MenuTopic Editors





Advances in Anti-Cancer Drugs
Topic Information
Dear Colleagues,
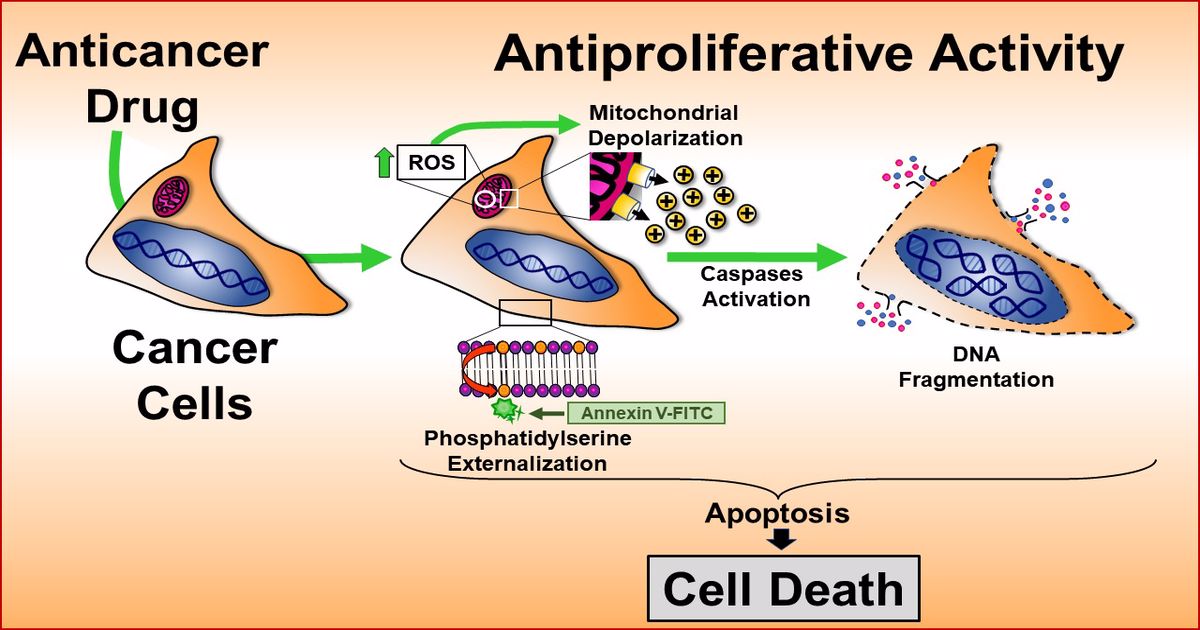
We hope this message finds you well. As large quantities of novel synthetic and natural molecules continue to be generated or discovered, there is challenging to identify and characterize therapeutic agents with effective anti-cancer activity. The aim of this particular Topic, "Advances in Anti-Cancer Drugs”, pretends aims to collect a group of publications focused on novel chemical compounds with cytotoxic activity on cancer cells in vitro, in vivo, or both, and particularly those articles including novel biomarkers and target proteins with potential therapeutic properties. In addition, studies on drug repurposing, including approved, discontinued, and shelved drugs, with anti-cancer activity, are encouraged for submission. Moreover, immunotherapy, electrochemotherapy, gene therapy, and phytomedicine studies are highly welcome. The submitted manuscripts should include the partial mechanism used for the novel compounds or strategies to induce cell death. This Topic provides a suitable platform to disseminate anti-cancer discoveries at the bench and the bedside. Thus, we are delighted to invite you for an excellent opportunity to publish your manuscript in our journal.
Dr. Armando Varela-Ramirez
Dr. Elisa Robles-Escajeda
Dr. Blanca E. Ruiz-Medina
Dr. Patricia Talamás-Rohana
Dr. Rachid Skouta
Topic Editors
Keywords
- anticancer
- antiproliferation
- apoptosis
- biomarkers
- cell cycle
- cell signaling
- drug discovery
- immunotherapy, gene therapy
- phytomedicine
Participating Journals
| Journal Name | Impact Factor | CiteScore | Launched Year | First Decision (median) | APC |
|---|---|---|---|---|---|
|
Biology
|
3.5 | 7.4 | 2012 | 16.8 Days | CHF 2700 |
|
Cancers
|
4.4 | 8.8 | 2009 | 19.1 Days | CHF 2900 |
|
Cells
|
5.2 | 10.5 | 2012 | 15.5 Days | CHF 2700 |
|
Current Oncology
|
3.4 | 4.9 | 1994 | 22.8 Days | CHF 2200 |
|
Medical Sciences
|
4.4 | 8.7 | 2013 | 18.7 Days | CHF 1600 |
|
Medicines
|
- | - | 2014 | 27.7 Days | CHF 1400 |
|
Pharmaceuticals
|
4.8 | 7.7 | 2004 | 16 Days | CHF 2900 |
![]()
Preprints.org is a multidisciplinary platform offering a preprint service designed to facilitate the early sharing of your research. It supports and empowers your research journey from the very beginning.
MDPI Topics is collaborating with Preprints.org and has established a direct connection between MDPI journals and the platform. Authors are encouraged to take advantage of this opportunity by posting their preprints at Preprints.org prior to publication:
- Share your research immediately: disseminate your ideas prior to publication and establish priority for your work.
- Safeguard your intellectual contribution: Protect your ideas with a time-stamped preprint that serves as proof of your research timeline.
- Boost visibility and impact: Increase the reach and influence of your research by making it accessible to a global audience.
- Gain early feedback: Receive valuable input and insights from peers before submitting to a journal.
- Ensure broad indexing: Web of Science (Preprint Citation Index), Google Scholar, Crossref, SHARE, PrePubMed, Scilit and Europe PMC.
Related Topic
- Advances in Anti-Cancer Drugs: 2nd Edition (12 articles)