




Norbornene and Related Structures as Scaffolds in the Search for New Cancer Treatments


 , , , ,
, , , , 

Abstract
1. Introduction
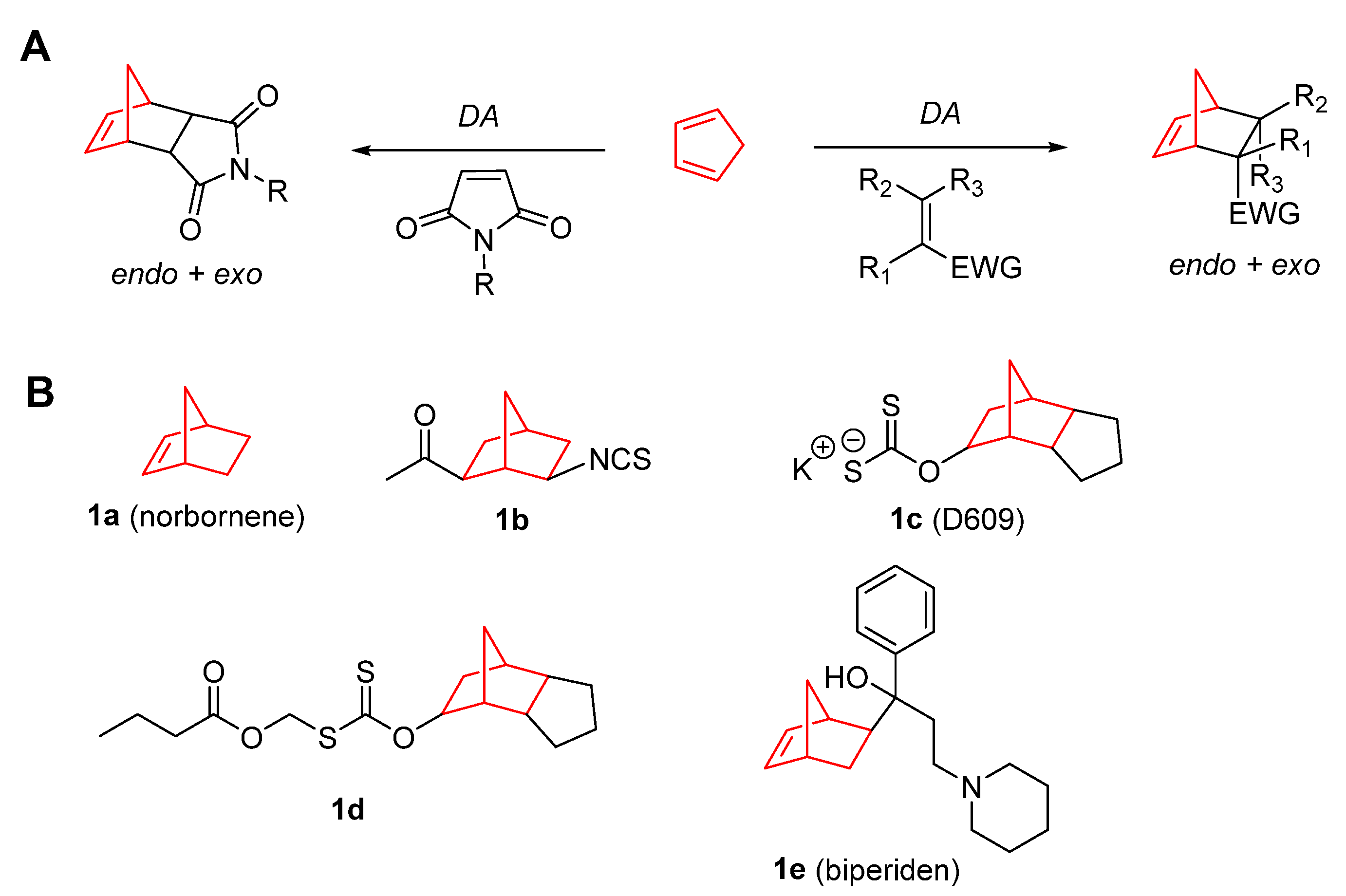
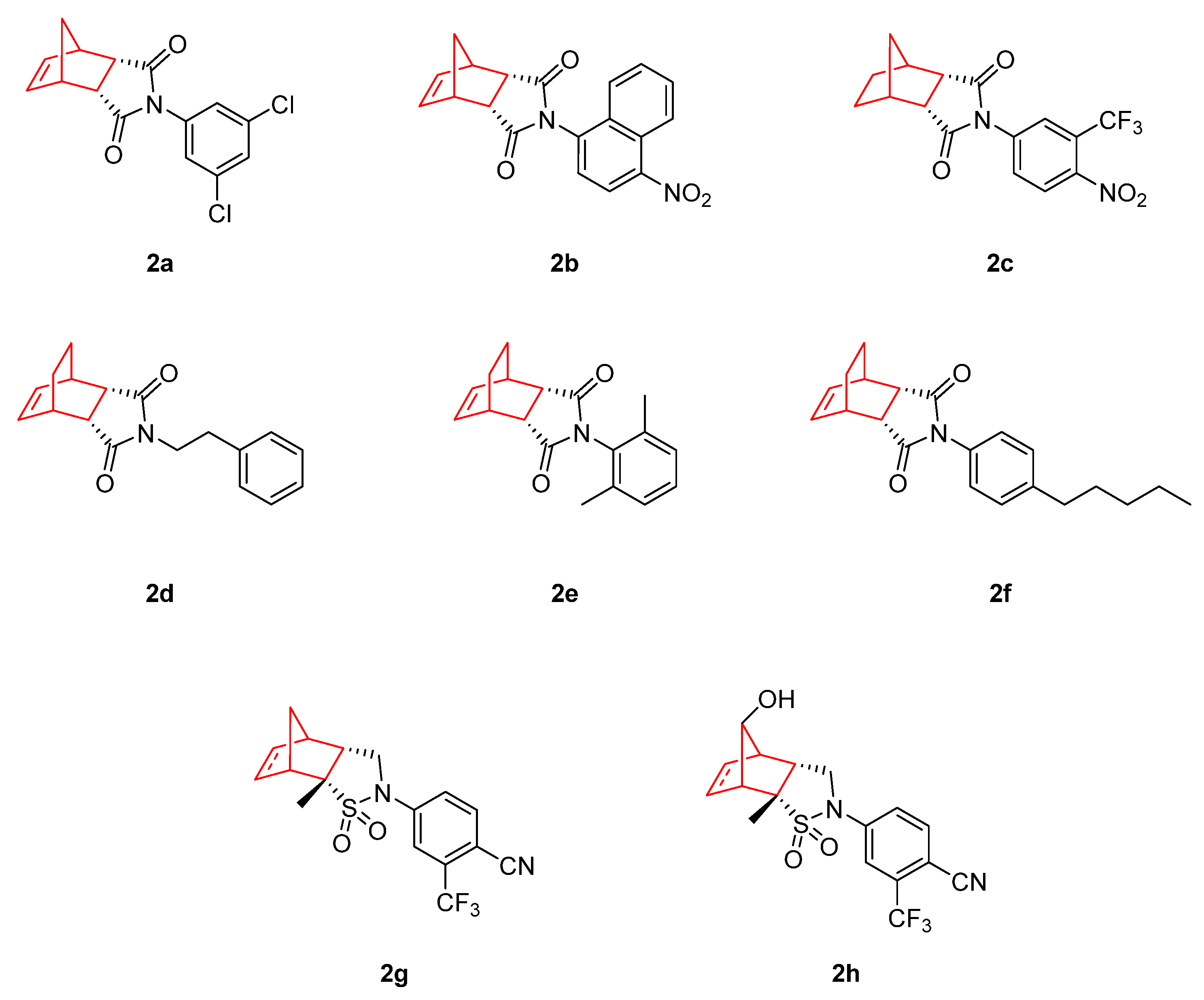
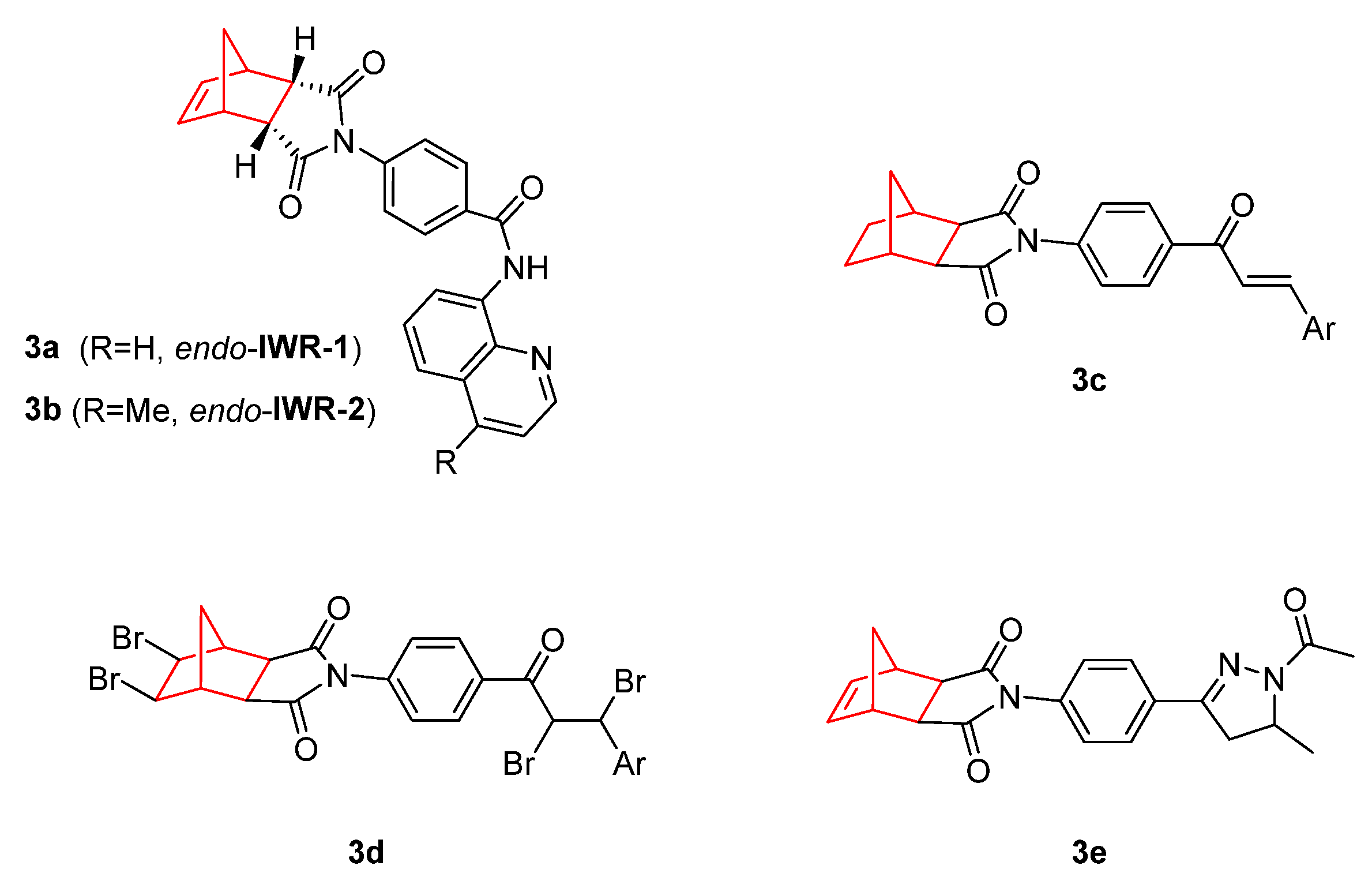
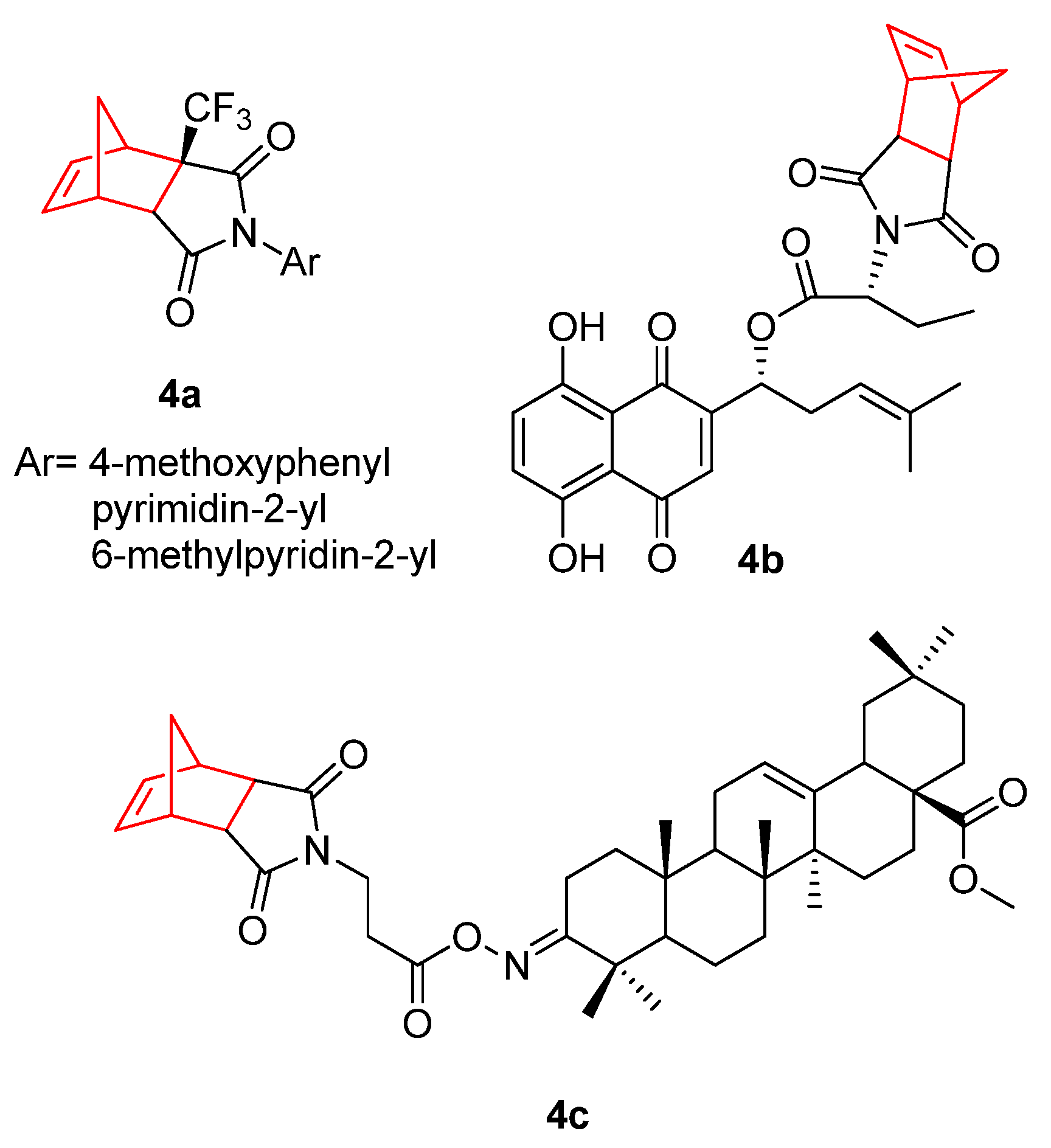
2. Therapeutic Potential of Norbornene Derivatives in Cancer
3. Camphor and Borneol Derivatives

4. Metal Complexes with Norbornene and Norbornene-Related Compounds
5. 7 Oxanorbornene Derivatives
6. Drug Delivery Systems Containing Norbornene and Related Scaffolds in the Treatment of Cancer
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ABCB1 | ABC transporters multidrug resistance 1 |
| ABCG2 | ABC transporters |
| ADMET | Chemical absorption, distribution, metabolism, excretion, and toxicity |
| ADT | Androgen deprivation therapy |
| Ag | Agonist |
| Akt | Serine/threonine kinase |
| ALK | Anaplastic lymphoma kinase |
| AMPK/mTOR | AMP-activated protein kinase/mechanistic target of rapamycin kinase |
| APC | Adenomatous polyposis coli gen |
| AR | Androgen receptor |
| AZA | Acetazolamide |
| BAX | BCL2 associated X protein |
| BBB | Blood–brain barrier |
| Bcl-XL | B-cell lymphoma-extra large |
| BTB | Blood–tumor barrier |
| BCL-2 | B-cell lymphoma 2 |
| BTCP | 2-bicyclo[2.2.1]hept-5-enyl-1H-1,3,7,8-tetraazacyclopenta[l]phenanthrene |
| CA | Carbonic anhydrase |
| cAMP | Cyclic adenosine monophosphate |
| CDK2 | Cyclin dependent kinase 2 |
| c-Met | Mesenchymal epithelial transition factor |
| DA | Diels–Alder |
| DDS | Drug delivery systems |
| DNA | Deoxyribonucleic acid |
| EC50 | Half maximal effective concentration |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial mesenchymal transmission |
| ER | Estrogen receptors |
| ERK | Estrogen receptor kinase |
| FAM46C | Non-canonical poly(A) polymerase protein |
| FDA | Food drug administration |
| HDAC | Histone deacetylase |
| HUVEC | Human Umbilical Vein Endothelial Cell |
| IARC | International Agency for Research on Cancer |
| IC50 | Half maximal inhibitory concentration |
| InR | Insulin receptor |
| JNK | c-Jun N-terminal kinase |
| Ki | Inhibitory constant |
| LC3-IIB | Microtubule-associated protein 1A/1B-light chain 3-phosphatidylethanolamine conjugate B |
| MALT1 | Mucosa-associated lymphoid tissue lymphoma translocation protein 1 |
| MAP | Mitogen-activated protein |
| MCL-1 | Myeloid cell leukemia-1 |
| MEK1/2 | Mitogen-activated protein kinase kinases 1/2 |
| MOF | Metal–organic framework |
| MT | Mutant type |
| PDAC | Pancreatic ductal adenocarcinoma |
| P-gp | MDR1/P-glycoprotein |
| Ras | Rat sarcoma virus |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| SDHB | Succinate dehydrogenase complex subunit b |
| SI | Selectivity Index |
| SIRT7 | Sirtuin protein 7 |
| SQSTM1/p62 | Sequestosome 1 |
| TGI | Total growth inhibition |
| TNF | Tumor necrosis factor |
| TRPM8 | Transient receptor potential cation channel subfamily M member 8 |
| VEGFR-2 | Vascular endothelial growth factor receptor 2 |
| WT | Wild type |
References
- World Health Organization Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 22 July 2022).
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; He, Z.; Liu, T.; Chen, D.; Wang, B.; Wen, Q.; Zheng, X. Evolution of Molecular Targeted Cancer Therapy: Mechanisms of Drug Resistance and Novel Opportunities Identified by CRISPR-Cas9 Screening. Front. Oncol. 2022, 12, 755053. [Google Scholar] [CrossRef] [PubMed]
- N, V.R.; Dinda, H.; Ganivada, M.N.; Das Sarma, J.; Shunmugam, R. Efficient Approach to Prepare Multiple Chemotherapeutic Agent Conjugated Nanocarrier. Chem. Commun. 2014, 50, 13540–13543. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dinda, H.; Chakraborty, I.; Bhattacharyya, R.; Das Sarma, J.; Shunmugam, R. Engineering Camptothecin-Derived Norbornene Polymers for Theranostic Application. ACS Omega 2017, 2, 2848–2857. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, G. A Convenient Preparation of Cyclopentadiene from Its Dimer. J. Org. Chem. 1985, 50, 1998. [Google Scholar] [CrossRef]
- Monson, R.S. The Diels-Alder Reaction. In Advanced Organic Synthesis; Elsevier: Amsterdam, The Netherlands, 1971; Volume 3, pp. 71–79. ISBN 047180343X. [Google Scholar]
- Huertas, D.; Florscher, M.; Dragojlovic, V. Solvent-Free Diels–Alder Reactions of in Situ Generated Cyclopentadiene. Green Chem. 2009, 11, 91–95. [Google Scholar] [CrossRef]
- Glassner, M.; Blinco, J.P.; Barner-Kowollik, C. Diels–Alder Reactions as an Efficient Route to High Purity Cyclic Polymers. Macromol. Rapid Commun. 2011, 32, 724–728. [Google Scholar] [CrossRef]
- Kaminsky, W.; Boggioni, L.; Tritto, I.; Aqida, S.N. Cycloolefin Polymerization. In Reference Module in Materials Science and Materials Engineering; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Hasan, T.; Ikeda, T.; Shiono, T. Ethene−Norbornene Copolymer with High Norbornene Content Produced by a Nsa -Fluorenylamidodimethyltitanium Complex Using a Suitable Activator. Macromolecules 2004, 37, 8503–8509. [Google Scholar] [CrossRef]
- Chowdhury, S.I.; Tanaka, R.; Nakayama, Y.; Shiono, T. Copolymerization of Norbornene and Styrene with Anilinonaphthoquinone-Ligated Nickel Complexes. Polymers 2019, 11, 1100. [Google Scholar] [CrossRef]
- Spanget-Larsen, J.; Gleiter, R. On the Exceptional Reactivity of the Norbornene Double Bond. Tetrahedron Lett. 1982, 23, 2435–2438. [Google Scholar] [CrossRef]
- Bartlett, P.D.; Ghosh, T. Sulfuration of the Norbornene Double Bond. J. Org. Chem. 1987, 52, 4937–4943. [Google Scholar] [CrossRef]
- Balaban, T.S.; Balaban, A.T. Five-Membered Rings with Three Oxygen or Sulfur Atoms in 1,2,4-Positions. In Comprehensive Heterocyclic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2008; Volume 6, pp. 191–256. ISBN 9780080449920. [Google Scholar]
- Lang, K.; Davis, L.; Torres-Kolbus, J.; Chou, C.; Deiters, A.; Chin, J.W. Genetically Encoded Norbornene Directs Site-Specific Cellular Protein Labelling via a Rapid Bioorthogonal Reaction. Nat. Chem. 2012, 4, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, B.L.; Guo, Z.; Bernardes, G.J.L. Inverse Electron Demand Diels–Alder Reactions in Chemical Biology. Chem. Soc. Rev. 2017, 46, 4895–4950. [Google Scholar] [CrossRef] [PubMed]
- Stockdale, T.P.; Williams, C.M. Pharmaceuticals That Contain Polycyclic Hydrocarbon Scaffolds. Chem. Soc. Rev. 2015, 44, 7737–7763. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kensler, T.W.; Cho, C.G.; Posner, G.H.; Talalay, P. Anticarcinogenic Activities of Sulforaphane and Structurally Related Synthetic Norbornyl Isothiocyanates. Proc. Natl. Acad. Sci. USA 1994, 91, 3147–3150. [Google Scholar] [CrossRef]
- Pörn-Ares, M.I.; Chow, S.C.; Slotte, J.P.; Orrenius, S. Induction of Apoptosis and Potentiation of TNF- and Fas-Mediated Apoptosis in U937 Cells by the Xanthogenate Compound D609. Exp. Cell Res. 1997, 235, 48–54. [Google Scholar] [CrossRef]
- Du, C.; Zhao, Q.; Araki, S.; Zhang, S.; Miao, J. Apoptosis Mediated by Phosphatidylcholine-Specific Phospholipase C Is Associated with CAMP, P53 Level, and Cell-Cycle Distribution in Vascular Endothelial Cells. Endothelium 2003, 10, 141–147. [Google Scholar] [CrossRef]
- Milhas, D.; Andrieu-Abadie, N.; Levade, T.; Benoist, H.; Ségui, B. The Tricyclodecan-9-Yl-Xanthogenate D609 Triggers Ceramide Increase and Enhances FasL-Induced Caspase-Dependent and -Independent Cell Death in T Lymphocytes. Int. J. Mol. Sci. 2012, 13, 8834–8852. [Google Scholar] [CrossRef]
- Kalluri, H.S.G.; Kuo, J.S.; Dempsey, R.J. Effect of D609 on the Expression of GADD45β Protein: Potential Inhibitory Role in the Growth of Glioblastoma Cancer Stem like Cells. Eur. J. Pharmacol. 2016, 791, 510–517. [Google Scholar] [CrossRef]
- Kalluri, H.S.G.; Kuo, J.S.; Dempsey, R.J. Chronic D609 Treatment Interferes with Cell Cycle and Targets the Expression of Olig2 in Glioma Stem like Cells. Eur. J. Pharmacol. 2017, 814, 81–86. [Google Scholar] [CrossRef]
- Bai, A.; Meier, G.P.; Wang, Y.; Luberto, C.; Hannun, Y.A.; Zhou, D. Prodrug Modification Increases Potassium Tricyclo[5.2.1.0 2,6 ]-Decan-8-Yl Dithiocarbonate (D609) Chemical Stability and Cytotoxicity against U937 Leukemia Cells. J. Pharmacol. Exp. Ther. 2004, 309, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Kostelnik, A.; Cegan, A.; Pohanka, M. Anti-Parkinson Drug Biperiden Inhibits Enzyme Acetylcholinesterase. Biomed Res. Int. 2017, 2017, 2532764. [Google Scholar] [CrossRef] [PubMed]
- Konczalla, L.; Perez, D.R.; Wenzel, N.; Wolters-Eisfeld, G.; Klemp, C.; Lüddeke, J.; Wolski, A.; Landschulze, D.; Meier, C.; Buchholz, A.; et al. Biperiden and Mepazine Effectively Inhibit MALT1 Activity and Tumor Growth in Pancreatic Cancer. Int. J. Cancer 2020, 146, 1618–1630. [Google Scholar] [CrossRef] [PubMed]
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocr. Rev. 2004, 25, 276–308. [Google Scholar] [CrossRef]
- Salvati, M.E.; Balog, A.; Shan, W.; Wei, D.D.; Pickering, D.; Attar, R.M.; Geng, J.; Rizzo, C.A.; Gottardis, M.M.; Weinmann, R.; et al. Structure Based Approach to the Design of Bicyclic-1H-Isoindole-1,3(2H)-Dione Based Androgen Receptor Antagonists. Bioorg. Med. Chem. Lett. 2005, 15, 271–276. [Google Scholar] [CrossRef]
- Salvati, M.E.; Balog, A.; Wei, D.D.; Pickering, D.; Attar, R.M.; Geng, J.; Rizzo, C.A.; Hunt, J.T.; Gottardis, M.M.; Weinmann, R.; et al. Identification of a Novel Class of Androgen Receptor Antagonists Based on the Bicyclic-1H-Isoindole-1,3(2H)-Dione Nucleus. Bioorg. Med. Chem. Lett. 2005, 15, 389–393. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M. Novel and Versatile Methodology for Synthesis of Cyclic Imides and Evaluation of Their Cytotoxic, DNA Binding, Apoptotic Inducing Activities and Molecular Modeling Study. Eur. J. Med. Chem. 2007, 42, 614–626. [Google Scholar] [CrossRef]
- Shan, W.; Balog, A.; Nation, A.; Zhu, X.; Chen, J.; Cvijic, M.E.; Geng, J.; Rizzo, C.A.; Spires, T.; Attar, R.M.; et al. [2.2.1]-Bicyclic Sultams as Potent Androgen Receptor Antagonists. Bioorg. Med. Chem. Lett. 2016, 26, 5707–5711. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Chen, B.; Dodge, M.E.; Tang, W.; Lu, J.; Ma, Z.; Fan, C.-W.; Wei, S.; Hao, W.; Kilgore, J.; Williams, N.S.; et al. Small Molecule–Mediated Disruption of Wnt-Dependent Signaling in Tissue Regeneration and Cancer. Nat. Chem. Biol. 2009, 5, 100–107. [Google Scholar] [CrossRef]
- Lu, J.; Ma, Z.; Hsieh, J.-C.; Fan, C.-W.; Chen, B.; Longgood, J.C.; Williams, N.S.; Amatruda, J.F.; Lum, L.; Chen, C. Structure–Activity Relationship Studies of Small-Molecule Inhibitors of Wnt Response. Bioorg. Med. Chem. Lett. 2009, 19, 3825–3827. [Google Scholar] [CrossRef]
- Lee, S.C.; Kim, O.-H.; Lee, S.K.; Kim, S.-J. IWR-1 Inhibits Epithelial-Mesenchymal Transition of Colorectal Cancer Cells through Suppressing Wnt/β-Catenin Signaling as Well as Survivin Expression. Oncotarget 2015, 6, 27146–27159. [Google Scholar] [CrossRef]
- Martins-Neves, S.R.; Paiva-Oliveira, D.I.; Fontes-Ribeiro, C.; Bovée, J.V.M.G.; Cleton-Jansen, A.-M.; Gomes, C.M.F. IWR-1, a Tankyrase Inhibitor, Attenuates Wnt/β-Catenin Signaling in Cancer Stem-like Cells and Inhibits in Vivo the Growth of a Subcutaneous Human Osteosarcoma Xenograft. Cancer Lett. 2018, 414, 1–15. [Google Scholar] [CrossRef]
- Narwal, M.; Venkannagari, H.; Lehtiö, L. Structural Basis of Selective Inhibition of Human Tankyrases. J. Med. Chem. 2012, 55, 1360–1367. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Budak, Y.; Gürdere, M.B.; Tekin, Ş.; Köprülü, T.K.; Ertürk, F.; Özcan, K.; Gülçin, İ.; Ceylan, M. Synthesis, Characterization, Anticancer, Antimicrobial and Carbonic Anhydrase Inhibition Profiles of Novel (3aR,4S,7R,7aS)-2-(4-((E)-3-(3-Aryl)Acryloyl)Phenyl)-3a,4,7,7a-Tetrahydro-1H-4,7-Methanoisoindole-1,3(2H)-Dione Derivatives. Bioorg. Chem. 2017, 70, 118–125. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Budak, Y.; Eligüzel, F.; Taslimi, P.; Kılıç, D.; Gulçin, İ.; Ceylan, M. Synthesis and Carbonic Anhydrase Inhibition of Tetrabromo Chalcone Derivatives. Arch. Pharm. 2017, 350, 1700198. [Google Scholar] [CrossRef]
- Kocyigit, U.M.; Budak, Y.; Gürdere, M.B.; Dürü, N.; Taslimi, P.; Gülçin, İ.; Ceylan, M. Synthesis and Investigation of Anticancer, Antibacterial Activities and Carbonic Anhydrase, Acetylcholinesterase Inhibition Profiles of Novel (3aR,4S,7R,7aS)-2-[4-[1-Acetyl-5-(Aryl/Heteroaryl)-4,5-Dihydro-1H-Pyrazol-3-Yl]Phenyl]-3a,4,7,7a-Tetrahydro-1H-4. Mon. Chem.-Chem. Mon. 2019, 150, 721–731. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef]
- Luzina, E.L.; Popov, A.V. Synthesis and Anticancer Activity Evaluation of 3,4-Mono- and Bicyclosubstituted N-(Het)Aryl Trifluoromethyl Succinimides. J. Fluor. Chem. 2014, 168, 121–127. [Google Scholar] [CrossRef]
- Baloch, S.K.; Ma, L.; Wang, X.-L.; Shi, J.; Zhu, Y.; Wu, F.-Y.; Pang, Y.-J.; Lu, G.-H.; Qi, J.-L.; Wang, X.-M.; et al. Design, Synthesis and Mechanism of Novel Shikonin Derivatives as Potent Anticancer Agents. RSC Adv. 2015, 5, 31759–31767. [Google Scholar] [CrossRef]
- Žiberna, L.; Šamec, D.; Mocan, A.; Nabavi, S.; Bishayee, A.; Farooqi, A.; Sureda, A.; Nabavi, S. Oleanolic Acid Alters Multiple Cell Signaling Pathways: Implication in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2017, 18, 643. [Google Scholar] [CrossRef]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Baltierra-Uribe, S.L.; Castillo-Cruz, J.; Campillo-Navarro, M.; Hernández-Pérez, A.D.; Luna-Angulo, A.B.; Chacón-Salinas, R.; Coral-Vázquez, R.M.; Estrada-García, I.; et al. Ursolic and Oleanolic Acids Induce Mitophagy in A549 Human Lung Cancer Cells. Molecules 2019, 24, 3444. [Google Scholar] [CrossRef]
- Ayeleso, T.; Matumba, M.; Mukwevho, E. Oleanolic Acid and Its Derivatives: Biological Activities and Therapeutic Potential in Chronic Diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef]
- Bednarczyk-Cwynar, B.; Ruszkowskp, P.; Atamanyuk, D.; Lesyk, R.; Zaprutko, L. Hybrids of Oleanolic Acid with Norbornene-2,3-Dicarboximide-N-Carboxylic Acids as Potential Anticancer Agents. Acta Pol. Pharm. 2017, 74, 827–835. [Google Scholar]
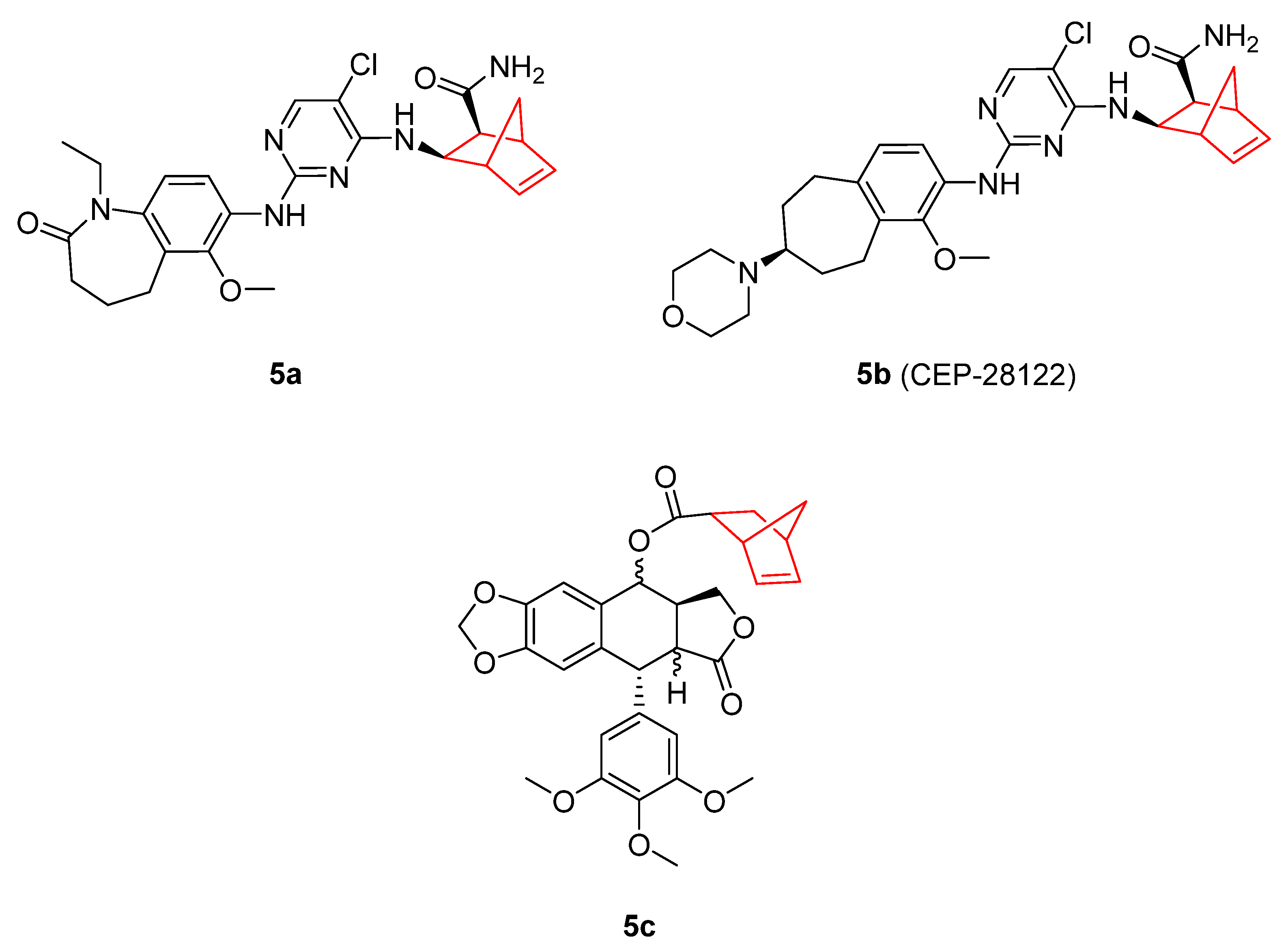
- Ott, G.R.; Tripathy, R.; Cheng, M.; McHugh, R.; Anzalone, A.V.; Underiner, T.L.; Curry, M.A.; Quail, M.R.; Lu, L.; Wan, W.; et al. Discovery of a Potent Inhibitor of Anaplastic Lymphoma Kinase with in Vivo Antitumor Activity. ACS Med. Chem. Lett. 2010, 1, 493–498. [Google Scholar] [CrossRef]
- Gingrich, D.E.; Lisko, J.G.; Curry, M.A.; Cheng, M.; Quail, M.; Lu, L.; Wan, W.; Albom, M.S.; Angeles, T.S.; Aimone, L.D.; et al. Discovery of an Orally Efficacious Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2012, 55, 4580–4593. [Google Scholar] [CrossRef]
- Cheng, M.; Quail, M.R.; Gingrich, D.E.; Ott, G.R.; Lu, L.; Wan, W.; Albom, M.S.; Angeles, T.S.; Aimone, L.D.; Cristofani, F.; et al. CEP-28122, a Highly Potent and Selective Orally Active Inhibitor of Anaplastic Lymphoma Kinase with Antitumor Activity in Experimental Models of Human Cancers. Mol. Cancer Ther. 2012, 11, 670–679. [Google Scholar] [CrossRef]
- López-Pérez, J.L.; del Olmo, E.; de Pascual-Teresa, B.; Abad, A.; San Feliciano, A. Synthesis and Cytotoxicity of Hydrophobic Esters of Podophyllotoxins. Bioorg. Med. Chem. Lett. 2004, 14, 1283–1286. [Google Scholar] [CrossRef]
- Zefirov, N.A.; Kruth, A.; Wobith, B.; Nurieva, E.V.; Riyaz, S.; Reddy, C.V.R.; Kuznetsov, S.A.; Zefirova, O.N. Novel Bridged and Caged C4-Podophyllotoxin Derivatives as Microtubule Disruptors: Synthesis, Cytotoxic Evaluation and Structure–Activity Relationship. Mendeleev Commun. 2018, 28, 475–478. [Google Scholar] [CrossRef]
- Itani, W.S.; El-Banna, S.H.; Hassan, S.B.; Larsson, R.L.; Bazarbachi, A.; Gali-Muhtasib, H.U. Anti Colon Cancer Components from Lebanese Sage (Salvia Libanotica) Essential Oil: Mechanistic Basis. Cancer Biol. Ther. 2008, 7, 1765–1773. [Google Scholar] [CrossRef]
- Moayedi, Y.; Greenberg, S.A.; Jenkins, B.A.; Marshall, K.L.; Dimitrov, L.V.; Nelson, A.M.; Owens, D.M.; Lumpkin, E.A. Camphor White Oil Induces Tumor Regression through Cytotoxic T Cell-dependent Mechanisms. Mol. Carcinog. 2019, 58, 722–734. [Google Scholar] [CrossRef] [PubMed]
- El Yaagoubi, M.; Ortiz, S.; Mechqoq, H.; Cavaleiro, C.; Lecsö-Bornet, M.; Rodrigues, M.J.; Custódio, L.; El Mousadik, A.; Grougnet, R.; El Aouad, N.; et al. Chemical Composition, Antibacterial Screening and Cytotoxic Activity of Chiliadenus Antiatlanticus (Asteraceae) Essential Oil. Chem. Biodivers. 2021, 18, e2100115. [Google Scholar] [CrossRef] [PubMed]
- Ouakouak, H.; Benarfa, A.; Messaoudi, M.; Begaa, S.; Sawicka, B.; Benchikha, N.; Simal-Gandara, J. Biological Properties of Essential Oils from Thymus Algeriensis Boiss. Plants 2021, 10, 786. [Google Scholar] [CrossRef]
- Mueller, S.O.; Kling, M.; Arifin Firzani, P.; Mecky, A.; Duranti, E.; Shields-Botella, J.; Delansorne, R.; Broschard, T.; Kramer, P.-J. Activation of Estrogen Receptor α and ERβ by 4-Methylbenzylidene-Camphor in Human and Rat Cells: Comparison with Phyto- and Xenoestrogens. Toxicol. Lett. 2003, 142, 89–101. [Google Scholar] [CrossRef]
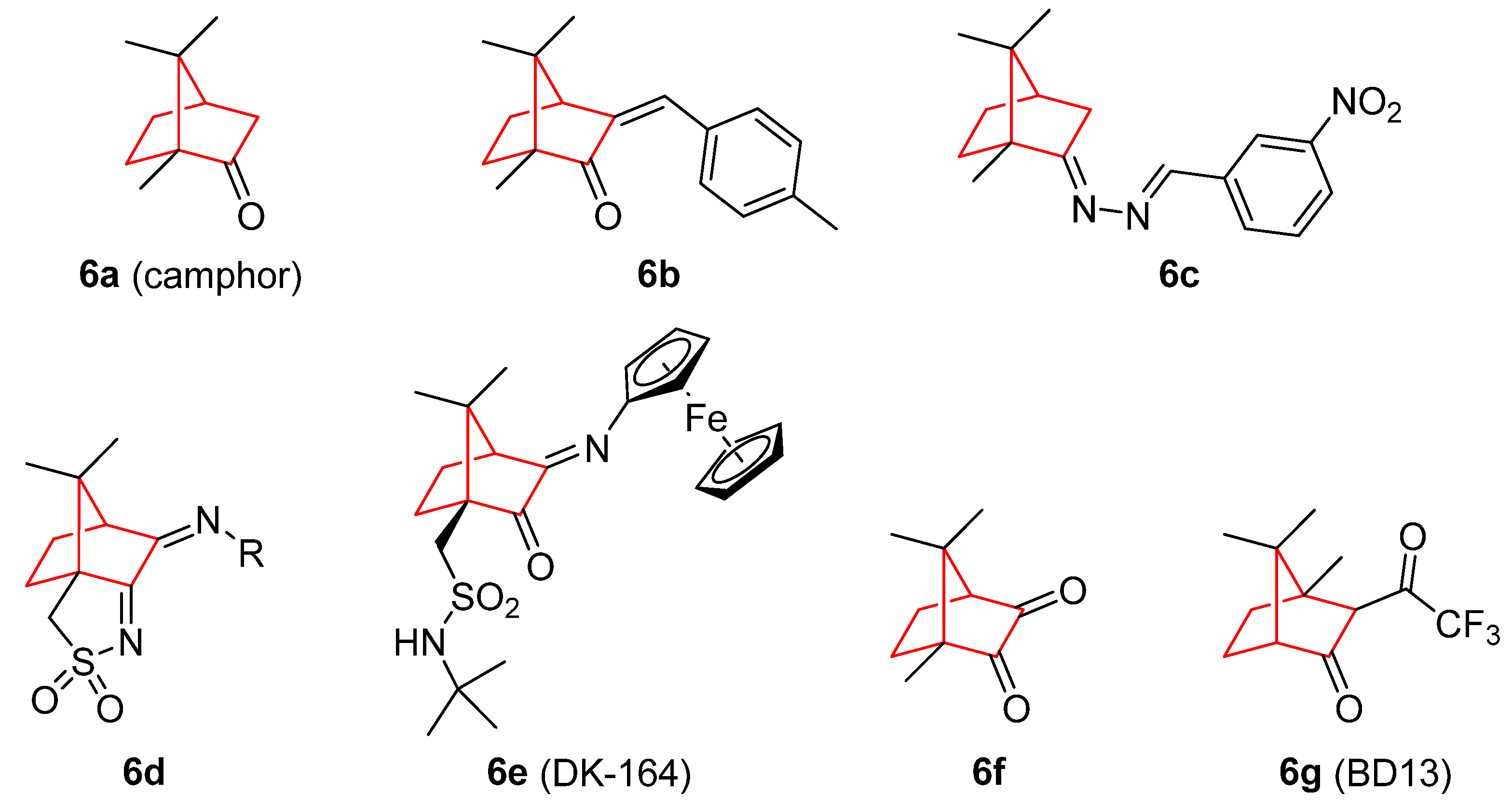
- da Silva, E.; da Silva Araújo, A.; Moraes, A.; de Souza, L.; Silva Lourenço, M.; de Souza, M.; Wardell, J.; Wardell, S. Synthesis and Biological Activities of Camphor Hydrazone and Imine Derivatives. Sci. Pharm. 2015, 84, 467–483. [Google Scholar] [CrossRef]
- Cardoso, J.M.S.; Correia, I.; Galvão, A.M.; Marques, F.; Carvalho, M.F.N.N. Synthesis of Ag(I) Camphor Sulphonylimine Complexes and Assessment of Their Cytotoxic Properties against Cisplatin -Resistant A2780cisR and A2780 Cell Lines. J. Inorg. Biochem. 2017, 166, 55–63. [Google Scholar] [CrossRef]
- Schröder, M.; Yusein-Myashkova, S.; Petrova, M.; Dobrikov, G.; Kamenova-Nacheva, M.; Todorova, J.; Pasheva, E.; Ugrinova, I. The Effect of a Ferrocene Containing Camphor Sulfonamide DK-164 on Breast Cancer Cell Lines. Anticancer. Agents Med. Chem. 2019, 19, 1874–1886. [Google Scholar] [CrossRef]
- Schröder, M.; Petrova, M.; Vlahova, Z.; Dobrikov, G.M.; Slavchev, I.; Pasheva, E.; Ugrinova, I. In Vitro Anticancer Activity of Two Ferrocene-Containing Camphor Sulfonamides as Promising Agents against Lung Cancer Cells. Biomedicines 2022, 10, 1353. [Google Scholar] [CrossRef]
- Volk, J.; Ziemann, C.; Leyhausen, G.; Geurtsen, W. Genotoxic and Mutagenic Potential of Camphorquinone in L5178/TK+/− Mouse Lymphoma Cells. Dent. Mater. 2018, 34, 519–530. [Google Scholar] [CrossRef]
- Nakano, K.; Nakayachi, T.; Yasumoto, E.; Morshed, S.R.M.D.; Hashimoto, K.; Kikuchi, H.; Nishikawa, H.; Sugiyama, K.; Amano, O.; Kawase, M.; et al. Induction of Apoptosis by Beta-Diketones in Human Tumor Cells. Anticancer Res. 2004, 24, 711–717. [Google Scholar]
- Verma, M.; Singh, S.; Bhushan, S.; Pal, H.; Kitchlu, S.; Koul, M.; Thappa, R.; Saxena, A. Induction of Mitochondrial-Dependent Apoptosis by an Essential Oil from Tanacetum Gracile. Planta Med. 2008, 74, 515–520. [Google Scholar] [CrossRef] [PubMed]
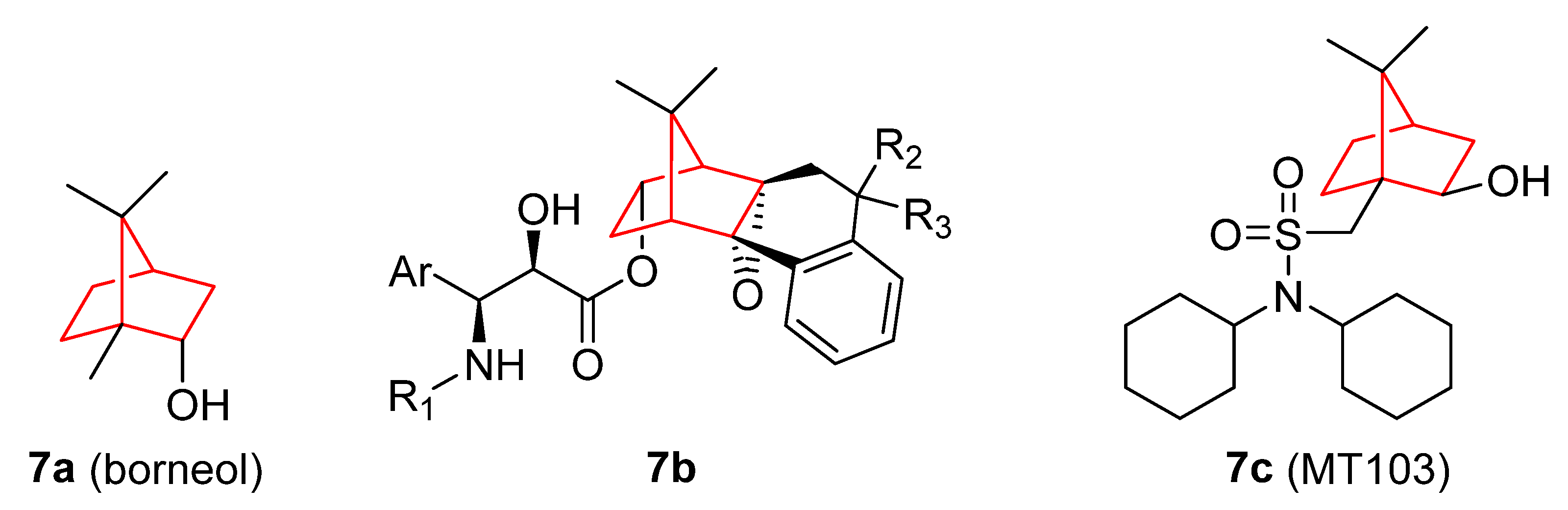
- Chen, J.; Li, L.; Su, J.; Li, B.; Chen, T.; Wong, Y.-S. Synergistic Apoptosis-Inducing Effects on A375 Human Melanoma Cells of Natural Borneol and Curcumin. PLoS ONE 2014, 9, e101277. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, L.; Su, J.; Chen, T. Natural Borneol Enhances Bisdemethoxycurcumin-Induced Cell Cycle Arrest in the G2/M Phase through up-Regulation of Intracellular ROS in HepG2 Cells. Food Funct. 2015, 6, 740–748. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, L.; Su, J.; Li, B.; Zhang, X.; Chen, T. Proteomic Analysis of G2/M Arrest Triggered by Natural Borneol/Curcumin in HepG2 Cells, the Importance of the Reactive Oxygen Species-P53 Pathway. J. Agric. Food Chem. 2015, 63, 6440–6449. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yin, Y.; Sun, J.; Feng, S.; Ma, J.; Fu, X.; Hou, Y.; Yang, M.; Sun, B.; Fan, C. Natural Borneol Is a Novel Chemosensitizer That Enhances Temozolomide-Induced Anticancer Efficiency against Human Glioma by Triggering Mitochondrial Dysfunction and Reactive Oxide Species-Mediated Oxidative Damage. Onco. Targets. Ther. 2018, 11, 5429–5439. [Google Scholar] [CrossRef]
- Meng, X.; Dong, X.; Wang, W.; Yang, L.; Zhang, X.; Li, Y.; Chen, T.; Ma, H.; Qi, D.; Su, J. Natural Borneol Enhances Paclitaxel-Induced Apoptosis of ESCC Cells by Inactivation of the PI3K/AKT. J. Food Sci. 2018, 83, 1436–1443. [Google Scholar] [CrossRef]
- Cao, W.; Li, Y.; Hou, Y.; Yang, M.; Fu, X.; Zhao, B.; Jiang, H.; Fu, X. Enhanced Anticancer Efficiency of Doxorubicin against Human Glioma by Natural Borneol through Triggering ROS-Mediated Signal. Biomed. Pharmacother. 2019, 118, 109261. [Google Scholar] [CrossRef]
- Lai, H.; Liu, C.; Hou, L.; Lin, W.; Chen, T.; Hong, A. TRPM8-Regulated Calcium Mobilization Plays a Critical Role in Synergistic Chemosensitization of Borneol on Doxorubicin. Theranostics 2020, 10, 10154–10170. [Google Scholar] [CrossRef]
- Duan, M.; Xing, Y.; Guo, J.; Chen, H.; Zhang, R. Borneol Increases Blood–Tumour Barrier Permeability by Regulating the Expression Levels of Tight Junction-Associated Proteins. Pharm. Biol. 2016, 54, 3009–3018. [Google Scholar] [CrossRef][Green Version]
- Chen, Z.; Xu, Q.; Shan, C.; Shi, Y.; Wang, Y.; Chang, R.C.-C.; Zheng, G. Borneol for Regulating the Permeability of the Blood-Brain Barrier in Experimental Ischemic Stroke: Preclinical Evidence and Possible Mechanism. Oxid. Med. Cell. Longev. 2019, 2019, 2936737. [Google Scholar] [CrossRef]
- Yin, Y.; Cao, L.; Ge, H.; Duanmu, W.; Tan, L.; Yuan, J.; Tunan, C.; Li, F.; Hu, R.; Gao, F.; et al. L-Borneol Induces Transient Opening of the Blood–Brain Barrier and Enhances the Therapeutic Effect of Cisplatin. Neuroreport 2017, 28, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Klar, U.; Graf, H.; Schenk, O.; Röhr, B.; Schulz, H. New Synthetic Inhibitors of Microtubule Depolymerization. Bioorg. Med. Chem. Lett. 1998, 8, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Jasinski, P.; Zwolak, P.; Isaksson Vogel, R.; Bodempudi, V.; Terai, K.; Galvez, J.; Land, D.; Dudek, A.Z. MT103 Inhibits Tumor Growth with Minimal Toxicity in Murine Model of Lung Carcinoma via Induction of Apoptosis. Investig. New Drugs 2011, 29, 846–852. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-Based Drugs: Past, Present and Future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Wang, G.; Yang, B.; Fu, Z.; Wang, X.; Zhang, Z. Efficacy and Safety of Oxaliplatin-Based Regimen versus Cisplatin-Based Regimen in the Treatment of Gastric Cancer: A Meta-Analysis of Randomized Controlled Trials. Int. J. Clin. Oncol. 2019, 24, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cisplatin versus Carboplatin: Comparative Review of Therapeutic Management in Solid Malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef] [PubMed]
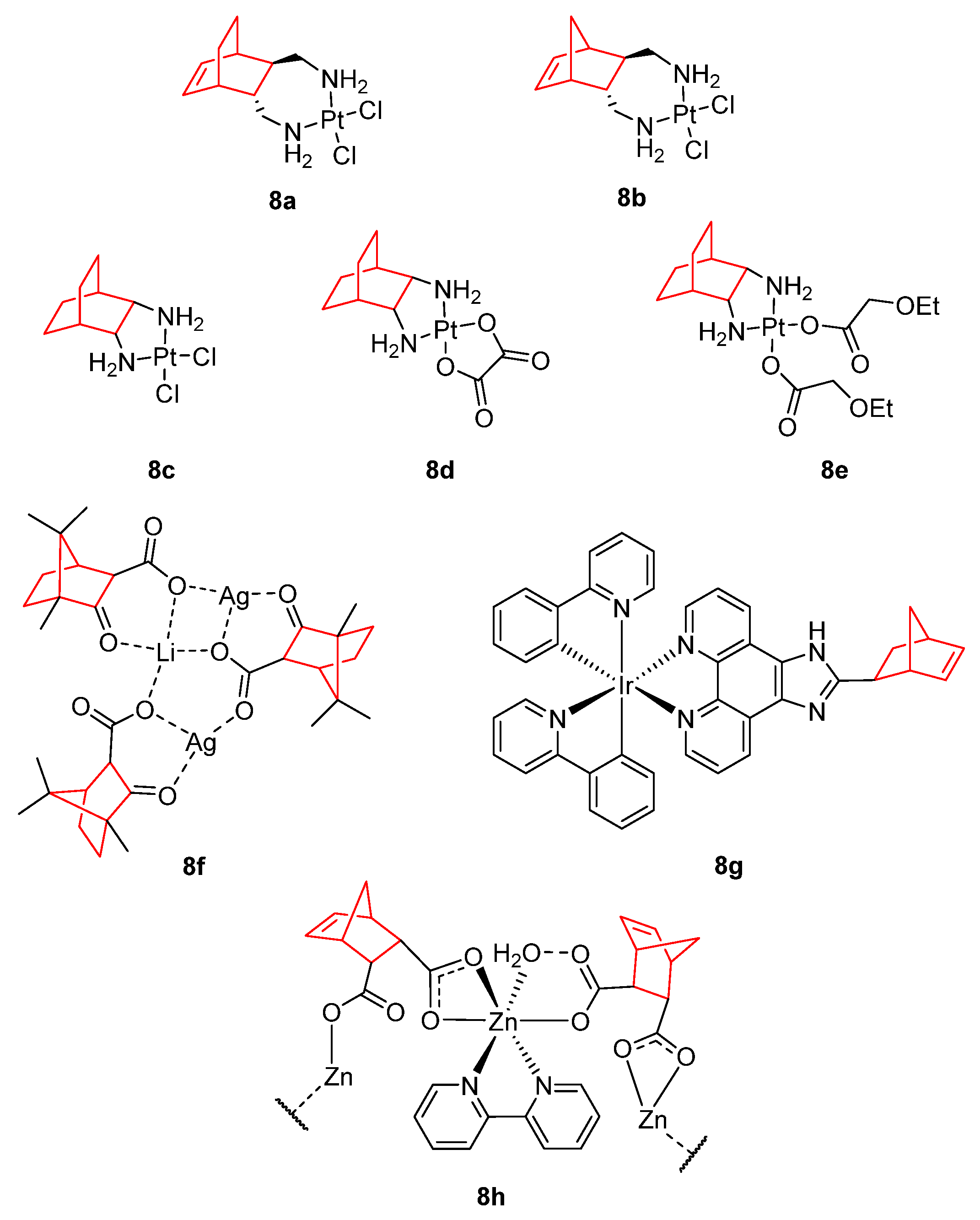
- de Mier-Vinué, J.; Gay, M.; Montaña, Á.M.; Sáez, R.-I.; Moreno, V.; Kasparkova, J.; Vrana, O.; Heringova, P.; Brabec, V.; Boccarelli, A.; et al. Synthesis, Biophysical Studies, and Antiproliferative Activity of Platinum(II) Complexes Having 1,2-Bis(Aminomethyl)Carbobicyclic Ligands. J. Med. Chem. 2008, 51, 424–431. [Google Scholar] [CrossRef]
- Liu, F.; Hu, W.; Fang, L.; Gou, S. Synthesis and Biological Evaluation of Water-Soluble Trans-[Bicyclo[2.2.2]Octane-7 R,8 R-Diamine]Platinum(II) Complexes with Linear or Branched Alkoxyacetates as Leaving Groups. J. Coord. Chem. 2016, 69, 1284–1292. [Google Scholar] [CrossRef]
- Liu, F.; Gou, S.; Chen, F.; Fang, L.; Zhao, J. Study on Antitumor Platinum(II) Complexes of Chiral Diamines with Dicyclic Species as Steric Hindrance. J. Med. Chem. 2015, 58, 6368–6377. [Google Scholar] [CrossRef]
- Carvalho, M.F.N.N.; Botelho do Rego, A.M.; Galvão, A.M.; Herrmann, R.; Marques, F. Search for Cytotoxic Compounds against Ovarian Cancer Cells: Synthesis, Characterization and Assessment of the Activity of New Camphor Carboxylate and Camphor Carboxamide Silver Complexes. J. Inorg. Biochem. 2018, 188, 88–95. [Google Scholar] [CrossRef]
- Zhang, C.; Lai, S.-H.; Zeng, C.-C.; Tang, B.; Wan, D.; Xing, D.-G.; Liu, Y.-J. The Induction of Apoptosis in SGC-7901 Cells through the ROS-Mediated Mitochondrial Dysfunction Pathway by a Ir(III) Complex. JBIC J. Biol. Inorg. Chem. 2016, 21, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Gao, E.; Sun, N.; Zhang, S.; Ding, Y.; Qiu, X.; Zhan, Y.; Zhu, M. Synthesis, Structures, Molecular Docking, Cytotoxicity and Bioimaging Studies of Two Novel Zn(II) Complexes. Eur. J. Med. Chem. 2016, 121, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jursic, B.S. Suitability of Furan, Pyrrole and Thiophene as Dienes for Diels–Alder Reactions Viewed through Their Stability and Reaction Barriers for Reactions with Acetylene, Ethylene and Cyclopropene. An AM1 Semiempirical and B3LYP Hybrid Density Functional Theory Study. J. Mol. Struct. THEOCHEM 1998, 454, 105–116. [Google Scholar] [CrossRef]
- McCluskey, A.; Ackland, S.P.; Bowyer, M.C.; Baldwin, M.L.; Garner, J.; Walkom, C.C.; Sakoff, J.A. Cantharidin Analogues: Synthesis and Evaluation of Growth Inhibition in a Panel of Selected Tumour Cell Lines. Bioorg. Chem. 2003, 31, 68–79. [Google Scholar] [CrossRef]
- Mo, L.; Zhang, X.; Shi, X.; Wei, L.; Zheng, D.; Li, H.; Gao, J.; Li, J.; Hu, Z. Norcantharidin Enhances Antitumor Immunity of GM-CSF Prostate Cancer Cells Vaccine by Inducing Apoptosis of Regulatory T Cells. Cancer Sci. 2018, 109, 2109–2118. [Google Scholar] [CrossRef]
- Yang, P.-Y.; Hu, D.-N.; Kao, Y.-H.; Lin, I.-C.; Chou, C.-Y.; Wu, Y.-C. Norcantharidin Induces Apoptosis in Human Prostate Cancer Cells through Both Intrinsic and Extrinsic Pathways. Pharmacol. Rep. 2016, 68, 874–880. [Google Scholar] [CrossRef]
- Peng, C.; Li, Z.; Niu, Z.; Niu, W.; Xu, Z.; Gao, H.; Niu, W.; Wang, J.; He, Z.; Gao, C.; et al. Norcantharidin Suppresses Colon Cancer Cell Epithelial-Mesenchymal Transition by Inhibiting the Avβ6-ERK-Ets1 Signaling Pathway. Sci. Rep. 2016, 6, 20500. [Google Scholar] [CrossRef]
- Pan, M.-S.; Cao, J.; Fan, Y.-Z. Insight into Norcantharidin, a Small-Molecule Synthetic Compound with Potential Multi-Target Anticancer Activities. Chin. Med. 2020, 15, 55. [Google Scholar] [CrossRef]
- Lin, C.-L.; Chen, C.-M.; Lin, C.-L.; Cheng, C.-W.; Lee, C.-H.; Hsieh, Y.-H. Norcantharidin Induces Mitochondrial-Dependent Apoptosis through Mcl-1 Inhibition in Human Prostate Cancer Cells. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1867–1876. [Google Scholar] [CrossRef]
- Zhou, J.; Ren, Y.; Tan, L.; Song, X.; Wang, M.; Li, Y.; Cao, Z.; Guo, C. Norcantharidin: Research Advances in Pharmaceutical Activities and Derivatives in Recent Years. Biomed. Pharmacother. 2020, 131, 110755. [Google Scholar] [CrossRef]
- Qiu, P.; Wang, S.; Liu, M.; Ma, H.; Zeng, X.; Zhang, M.; Xu, L.; Cui, Y.; Xu, H.; Tang, Y.; et al. Norcantharidin Inhibits Cell Growth by Suppressing the Expression and Phosphorylation of Both EGFR and C-Met in Human Colon Cancer Cells. BMC Cancer 2017, 17, 55. [Google Scholar] [CrossRef]
- Peng, C.; Liu, X.; Liu, E.; Xu, K.; Niu, W.; Chen, R.; Wang, J.; Zhang, Z.; Lin, P.; Wang, J.; et al. Norcantharidin Induces HT-29 Colon Cancer Cell Apoptosis through the Avβ6-Extracellular Signal-Related Kinase Signaling Pathway. Cancer Sci. 2009, 100, 2302–2308. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-L.; Cai, Y.-C.; Liu, X.-H.; Xian, L.-J. Norcantharidin Inhibits DNA Replication and Induces Apoptosis with the Cleavage of Initiation Protein Cdc6 in HL-60 Cells. Anticancer. Drugs 2006, 17, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.-Y.; Chen, M.-F.; Kao, Y.-H.; Hu, D.-N.; Chang, F.-R.; Wu, Y.-C. Norcantharidin Induces Apoptosis of Breast Cancer Cells: Involvement of Activities of Mitogen Activated Protein Kinases and Signal Transducers and Activators of Transcription. Toxicol. Vitr. 2011, 25, 699–707. [Google Scholar] [CrossRef] [PubMed]
- An, W.; Gong, X.; Wang, M.; Tashiro, S.; Onodera, S.; Ikejima, T. Norcantharidin Induces Apoptosis in HeLa Cells through Caspase, MAPK, and Mitochondrial Pathways. Acta Pharmacol. Sin. 2004, 25, 1502–1508. [Google Scholar]
- Zhang, Q.-Y.; Yue, X.-Q.; Jiang, Y.-P.; Han, T.; Xin, H.-L. FAM46C Is Critical for the Anti-Proliferation and pro-Apoptotic Effects of Norcantharidin in Hepatocellular Carcinoma Cells. Sci. Rep. 2017, 7, 396. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, Y.; Hua, Y.; Hu, C.; Zhong, Y. NCTD Elicits Proapoptotic and Antiglycolytic Effects on Colorectal Cancer Cells via Modulation of Fam46c Expression and Inhibition of ERK1/2 Signaling. Mol. Med. Rep. 2020, 22, 774–782. [Google Scholar] [CrossRef]
- Chen, A.W.-G.; Tseng, Y.-S.; Lin, C.-C.; Hsi, Y.-T.; Lo, Y.-S.; Chuang, Y.-C.; Lin, S.-H.; Yu, C.-Y.; Hsieh, M.-J.; Chen, M.-K. Norcantharidin Induce Apoptosis in Human Nasopharyngeal Carcinoma through Caspase and Mitochondrial Pathway. Environ. Toxicol. 2018, 33, 343–350. [Google Scholar] [CrossRef]
- Zhu, Y.; Mi, Y.; Wang, Z.; Jia, X.; Jin, Z. Norcantharidin Inhibits Viability and Induces Cell Cycle Arrest and Apoptosis in Osteosarcoma. Oncol. Lett. 2018, 17, 456–461. [Google Scholar] [CrossRef]
- He, Q.; Xue, S.; Tan, Y.; Zhang, L.; Shao, Q.; Xing, L.; Li, Y.; Xiang, T.; Luo, X.; Ren, G. Dual Inhibition of Akt and ERK Signaling Induces Cell Senescence in Triple-Negative Breast Cancer. Cancer Lett. 2019, 448, 94–104. [Google Scholar] [CrossRef]
- Liu, Z.; Li, B.; Cao, M.; Jiang, J. Norcantharidin Triggers Apoptotic Cell Death in Non-Small Cell Lung Cancer via a Mitophagy-Mediated Autophagy Pathway. Ann. Transl. Med. 2021, 9, 971. [Google Scholar] [CrossRef]
- Ahn, C.-H.; Hong, K.-O.; Jin, B.; Lee, W.; Jung, Y.C.; Lee, H.; Shin, J.-A.; Cho, S.-D.; Hong, S.D. Contribution of P38 MAPK Pathway to Norcantharidin-Induced Programmed Cell Death in Human Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 3487. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tuergong, G.; Zhu, H.; Wang, X.; Weng, G.; Ren, Y. Norcantharidin Induces G2/M Arrest and Apoptosis via Activation of ERK and JNK, but Not P38 Signaling in Human Renal Cell Carcinoma ACHN Cells. Acta Pharm. 2021, 71, 267–278. [Google Scholar] [CrossRef]
- Wu, J.-Y.; Kuo, C.-D.; Chu, C.-Y.; Chen, M.-S.; Lin, J.-H.; Chen, Y.-J.; Liao, H.-F. Synthesis of Novel Lipophilic N-Substituted Norcantharimide Derivatives and Evaluation of Their Anticancer Activities. Molecules 2014, 19, 6911–6928. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Wu, J.-Y.; Liao, H.-F.; Chen, Y.-J.; Kuo, C.-D. Comparative Assessment of Therapeutic Safety of Norcantharidin, N-Farnesyloxy-Norcantharimide, and N-Farnesyl-Norcantharimide against Jurkat T Cells Relative to Human Normal Lymphoblast. Medicine 2016, 95, e4467. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-S.; Shi, Y.; Ma, X.-N.; Xing, D.-X.; Liu, L.-D.; Liu, Y.; Zhao, Y.-X.; Sui, Q.-C.; Tan, X.-J. Synthesis, Crystal Structure, Spectroscopic Properties and Potential Anti-Cancerous Activities of Four Unsaturated Bis-Norcantharimides. J. Mol. Struct. 2016, 1115, 228–240. [Google Scholar] [CrossRef]
- Spare, L.K.; Falsetta, P.; Gilbert, J.; Harman, D.G.; Baker, M.A.; Li, F.; McCluskey, A.; Clegg, J.K.; Sakoff, J.A.; Aldrich-Wright, J.R.; et al. Cytotoxicity of a Series of Norcantharidin-Inspired Tetrahydroepoxyisoindole Carboxamides. ChemMedChem 2017, 12, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Huang, M.; Zhao, C.K.; Li, C.; Xu, L. Design, Synthesis, and Biological Activity Evaluation of Campthothecin-HAA-Norcantharidin Conjugates as Antitumor Agents in Vitro. Chem. Biol. Drug Des. 2019, 93, 986–992. [Google Scholar] [CrossRef]
- Zhao, C.K.; Li, C.; Wang, X.H.; Bao, Y.J.; Yang, F.H.; Huang, M. The Regio-Selective Synthesis of 10-Hydroxy Camptothecin Norcantharidin Conjugates and Their Biological Activity Evaluation in Vitro. R. Soc. Open Sci. 2018, 5, 172317. [Google Scholar] [CrossRef]
- Zhao, C.; Jia, J.; Wang, X.; Luo, C.; Wang, Y. Synthesis of Norcantharidin Complex Salts. J. Heterocycl. Chem. 2019, 56, 1567–1570. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Zhu, Y.; Li, X.-F.; Tang, L.-P.; Su, Z.-Q.; Wang, X.-Q.; Li, C.-Y.; Yang, H.-M.; Zheng, G.-J.; Feng, B. Norcantharidin Alone or in Combination with Crizotinib Induces Autophagic Cell Death in Hepatocellular Carcinoma by Repressing C-Met-MTOR Signaling. Oncotarget 2017, 8, 114945–114955. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-P.; Luo, L.-L.; Liu, Y.-Q.; Liu, X.-S.; An, F.-Y.; Sun, S.-B.; Xie, X.-R.; Geng, G.-Q.; Chen, X.-J.; Li, Z.-D. Norcantharidin Combined with Diamminedichloroplatinum Inhibits Tumor Growth and Cancerometastasis of Hepatic Carcinoma in Murine. J. Cancer Res. Ther. 2018, 14, 1035. [Google Scholar] [CrossRef]
- Singh, R.; Cheng, S.; Li, J.; Kumar, S.; Zeng, Q.; Zeng, Q. Norcantharidin Combined with 2-Deoxy-d-Glucose Suppresses the Hepatocellular Carcinoma Cells Proliferation and Migration. 3 Biotech 2021, 11, 142. [Google Scholar] [CrossRef]
- Wu, M.; Hui, S.; Chen, Y.; Chiou, H.; Lin, C.; Lee, C.; Hsieh, Y. Norcantharidin Combined with Paclitaxel Induces Endoplasmic Reticulum Stress Mediated Apoptotic Effect in Prostate Cancer Cells by Targeting SIRT7 Expression. Environ. Toxicol. 2021, 36, 2206–2216. [Google Scholar] [CrossRef]
- Wang, G.-S. Medical Uses of Mylabris in Ancient China and Recent Studies. J. Ethnopharmacol. 1989, 26, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xia, Z.; Chen, Y.; Yang, F.; Feng, W.; Cai, H.; Mei, Y.; Jiang, Y.; Xu, K.; Feng, D. Cantharidin Inhibits the Growth of Triple-Negative Breast Cancer Cells by Suppressing Autophagy and Inducing Apoptosis in Vitro and in Vivo. Cell. Physiol. Biochem. 2017, 43, 1829–1840. [Google Scholar] [CrossRef]
- Wang, W.-J.; Wu, M.-Y.; Shen, M.; Zhi, Q.; Liu, Z.-Y.; Gong, F.-R.; Tao, M.; Li, W. Cantharidin and Norcantharidin Impair Stemness of Pancreatic Cancer Cells by Repressing the β-Catenin Pathway and Strengthen the Cytotoxicity of Gemcitabine and Erlotinib. Int. J. Oncol. 2015, 47, 1912–1922. [Google Scholar] [CrossRef]
- Sakoff, J.A.; Ackland, S.P.; Baldwin, M.L.; Keane, M.A.; McCluskey, A. Anticancer Activity and Protein Phosphatase 1 and 2A Inhibition of a New Generation of Cantharidin Analogues. Investig. New Drugs 2002, 20, 1–11. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, S.; Zhang, H.; Gao, L.; Gao, X.; Liu, P.; Yi, Z.; Li, N.; Xu, Z. Combination Radiotherapy and Cantharidin Inhibits Lung Cancer Growth through Altering Tumor Infiltrating Lymphocytes. Futur. Oncol. 2017, 13, 1173–1180. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, L.; Thu, P.M.; Min, W.; Yang, P.; Li, J.; Li, P.; Xu, X. Sodium Cantharidinate, a Novel Anti-Pancreatic Cancer Agent That Activates Functional P53. Sci. China Life Sci. 2021, 64, 1295–1310. [Google Scholar] [CrossRef]
- Pang, J.; Huang, F.; Zhang, Y.; Wu, Y.; Ge, X.; Li, S.; Li, X. Sodium Cantharidate Induces Apoptosis in Breast Cancer Cells by Regulating Energy Metabolism via the Protein Phosphatase 5-P53 Axis. Toxicol. Appl. Pharmacol. 2021, 430, 115726. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, M.; Fan, W.; Yang, M.; Yang, L. Combination of Sodium Cantharidinate with Cisplatin Synergistically Hampers Growth of Cervical Cancer. Drug Des. Devel. Ther. 2021, 15, 171–183. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, P.; Lin, C.; Yang, W.; Ho, Y.; Yang, S.; Chuang, C. Cantharidic Acid Induces Apoptosis in Human Nasopharyngeal Carcinoma Cells through P38-mediated Upregulation of Caspase Activation. Environ. Toxicol. 2020, 35, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.D.; Mao, Y.; Dong, X.D.; Lei, Z.N.; Yang, Y.; Lin, L.; Ashby, C.R.; Yang, D.H.; Fan, Y.F.; Chen, Z.S. Methyl-Cantharidimide (MCA) Has Anticancer Efficacy in ABCB1- and ABCG2-Overexpressing and Cisplatin Resistant Cancer Cells. Front. Oncol. 2020, 10, 932. [Google Scholar] [CrossRef]
- Chiang, L.-L.; Tseng, I.-J.; Lin, P.-Y.; Sheu, S.-Y.; Lin, C.-T.; Hsieh, Y.-H.; Lin, Y.-J.; Chen, H.-L.; Lin, M.-H. Synthesis of Canthardin Sulfanilamides and Their Acid Anhydride Analogues via a Ring-Opening Reaction of Activated Aziridines and Their Associated Pharmacological Effects. Molecules 2016, 21, 100. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dong, J.; Deng, L. Overview of Cantharidin and Its Analogues. Curr. Med. Chem. 2018, 25, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-B.; Su, C.-J.; Hwang, J.-M.; Chou, M.-C. Therapeutic Effects of Cantharidin Analogues without Bridging Ether Oxygen on Human Hepatocellular Carcinoma Cells. Eur. J. Med. Chem. 2010, 45, 3981–3985. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.-B.; Comninos, J.S.; Stossi, F.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Synthesis and Evaluation of Estrogen Receptor Ligands with Bridged Oxabicyclic Cores Containing a Diarylethylene Motif: Estrogen Antagonists of Unusual Structure. J. Med. Chem. 2005, 48, 7261–7274. [Google Scholar] [CrossRef]
- Zhao, Y.; Gong, P.; Chen, Y.; Nwachukwu, J.C.; Srinivasan, S.; Ko, C.; Bagchi, M.K.; Taylor, R.N.; Korach, K.S.; Nettles, K.W.; et al. Dual Suppression of Estrogenic and Inflammatory Activities for Targeting of Endometriosis. Sci. Transl. Med. 2015, 7, 271ra9. [Google Scholar] [CrossRef]
- Wu, J.; Yan, J.; Fang, P.; Zhou, H.; Liang, K.; Huang, J. Three-Dimensional Oxabicycloheptene Sulfonate Targets the Homologous Recombination and Repair Programmes through Estrogen Receptor α Antagonism. Cancer Lett. 2020, 469, 78–88. [Google Scholar] [CrossRef]
- Tang, C.; Li, C.; Zhang, S.; Hu, Z.; Wu, J.; Dong, C.; Huang, J.; Zhou, H.-B. Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. J. Med. Chem. 2015, 58, 4550–4572. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, C.; Hu, Z.; Zhao, C.; Li, C.; Zhang, S.; Dong, C.; Zhou, H.-B.; Huang, J. Synthesis and Structure–Activity Relationships of Novel Hybrid Ferrocenyl Compounds Based on a Bicyclic Core Skeleton for Breast Cancer Therapy. Bioorg. Med. Chem. 2016, 24, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Ning, W.; Hu, Z.; Tang, C.; Yang, L.; Zhang, S.; Dong, C.; Huang, J.; Zhou, H.-B. Novel Hybrid Conjugates with Dual Suppression of Estrogenic and Inflammatory Activities Display Significantly Improved Potency against Breast Cancer. J. Med. Chem. 2018, 61, 8155–8173. [Google Scholar] [CrossRef]
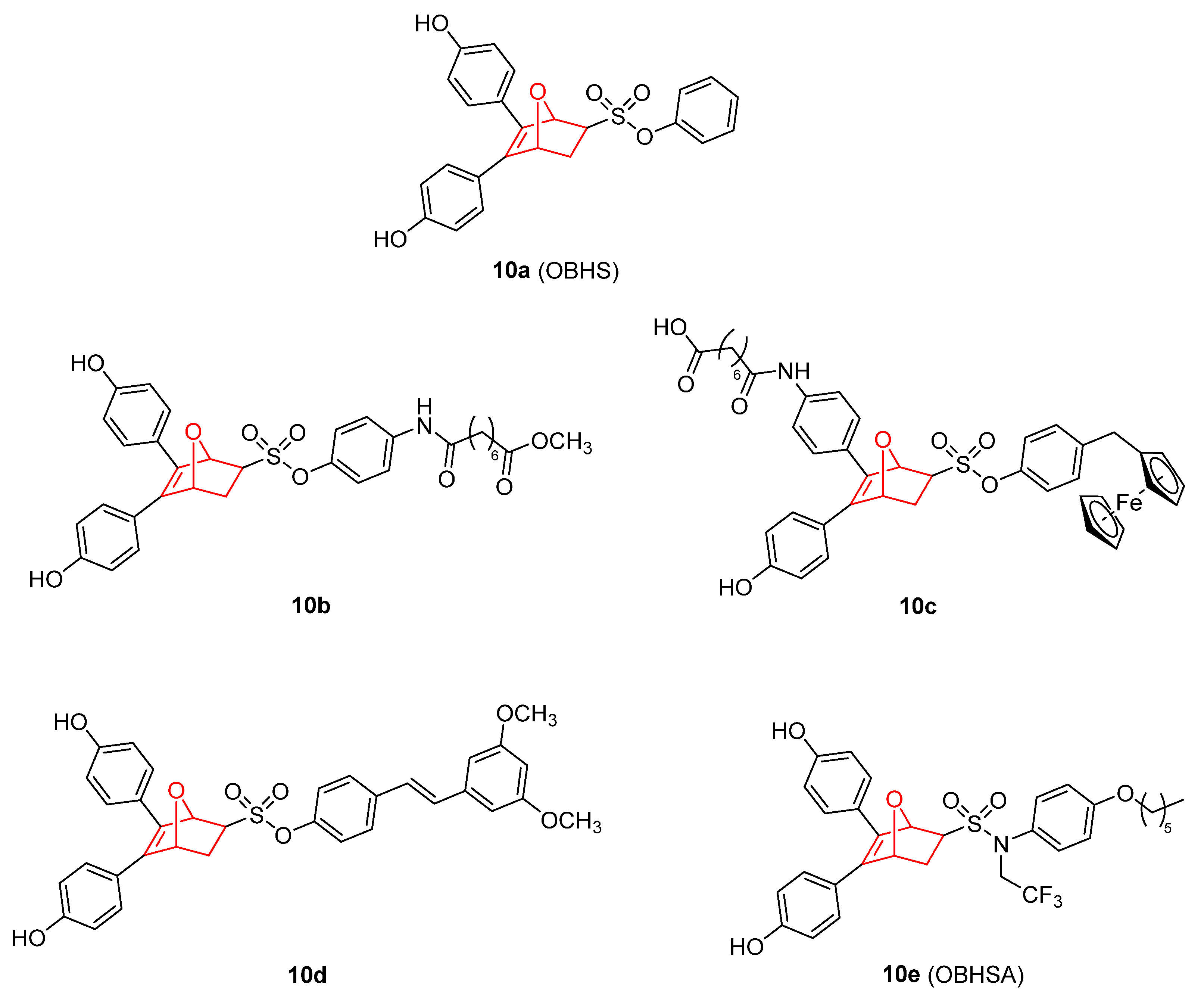
- Hu, Z.; Li, Y.; Xie, B.; Ning, W.; Xiao, Y.; Huang, Y.; Zhao, C.; Huang, J.; Dong, C.; Zhou, H.-B. Novel Class of 7-Oxabicyclo[2.2.1]Heptene Sulfonamides with Long Alkyl Chains Displaying Improved Estrogen Receptor α Degradation Activity. Eur. J. Med. Chem. 2019, 182, 111605. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, D.; Sancineto, L.; Fabro de Bem, A.; Tew, K.D.; Santi, C.; Radi, R.; Toquato, P.; Galli, F. Selenocompounds in Cancer Therapy: An Overview. Adv. Cancer Res. 2017, 136, 259–302. [Google Scholar] [PubMed]
- Ali, W.; Álvarez-Pérez, M.; Marć, M.A.; Salardón-Jiménez, N.; Handzlik, J.; Domínguez-Álvarez, E. The Anticancer and Chemopreventive Activity of Selenocyanate-Containing Compounds. Curr. Pharmacol. Rep. 2018, 4, 468–481. [Google Scholar] [CrossRef]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic Selenium Compounds as Potential Chemotherapeutic Agents for Improved Cancer Treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Garnica, P.; Encío, I.; Plano, D.; Palop, J.A.; Sanmartín, C. Organoseleno Cytostatic Derivatives: Autophagic Cell Death with AMPK and JNK Activation. Eur. J. Med. Chem. 2019, 175, 234–246. [Google Scholar] [CrossRef]
- Moradi Kashkooli, F.; Soltani, M.; Souri, M. Controlled Anti-Cancer Drug Release through Advanced Nano-Drug Delivery Systems: Static and Dynamic Targeting Strategies. J. Control. Release 2020, 327, 316–349. [Google Scholar] [CrossRef]
- Kulkarni, M.; Sawant, N.; Kolapkar, A.; Huprikar, A.; Desai, N. Borneol: A Promising Monoterpenoid in Enhancing Drug Delivery Across Various Physiological Barriers. AAPS PharmSciTech 2021, 22, 145. [Google Scholar] [CrossRef]
- Guo, X.; Wu, G.; Wang, H.; Chen, L. Pep-1&borneol–Bifunctionalized Carmustine-Loaded Micelles Enhance Anti-Glioma Efficacy Through Tumor-Targeting and BBB-Penetrating. J. Pharm. Sci. 2019, 108, 1726–1735. [Google Scholar] [CrossRef] [PubMed]
- Meng, L.; Chu, X.; Xing, H.; Liu, X.; Xin, X.; Chen, L.; Jin, M.; Guan, Y.; Huang, W.; Gao, Z. Improving Glioblastoma Therapeutic Outcomes via Doxorubicin-Loaded Nanomicelles Modified with Borneol. Int. J. Pharm. 2019, 567, 118485. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Guan, Q.-X.; Shang, Y.-Z.; Wang, Y.-H.; Lv, S.-W.; Yang, Z.-X.; Wang, R.; Feng, Y.-F.; Li, W.-N.; Li, Y.-J. Metal-Organic Framework IRMOFs Coated with a Temperature-Sensitive Gel Delivering Norcantharidin to Treat Liver Cancer. World J. Gastroenterol. 2021, 27, 4208–4220. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Jiang, Z.; Qiao, J.; Peng, Y.; Liu, W.; Han, B. Synthesis and Anti-Metastasis Activities of Norcantharidin-Conjugated Carboxymethyl Chitosan as a Novel Drug Delivery System. Carbohydr. Polym. 2019, 214, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Lei, M.; Shi, K.; Wang, M.; Hao, Y.; Xiao, Y.; Yuan, L.; Peng, J.; Qian, Z. Intratumoral Injection of Norcantharidin-Loaded Poly(D,L-Lactide)-b-Poly(Ethylene Glycol)-b-Poly(D,L-Lactide) Thermosensitive Hydrogel for the Treatment of Primary Hepatocellular Carcinoma. J. Biomed. Nanotechnol. 2019, 15, 2025–2044. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, X.; Li, H.; Yan, D.; Huang, W. Two Zn(II) Coordination Polymers with Anticancer Drug Norcantharidin as Ligands for Cancer Chemotherapy. Dalt. Trans. 2022, 51, 5624–5634. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tu, J.; Feng, Y.; Zhang, J.; Wu, J. Synergistic Co-Delivery of Diacid Metabolite of Norcantharidin and ABT-737 Based on Folate-Modified Lipid Bilayer-Coated Mesoporous Silica Nanoparticle against Hepatic Carcinoma. J. Nanobiotechnology 2020, 18, 114. [Google Scholar] [CrossRef]
- Zhou, L.; Zou, M.; Zhu, K.; Ning, S.; Xia, X. Development of 11-DGA-3-O-Gal-Modified Cantharidin Liposomes for Treatment of Hepatocellular Carcinoma. Molecules 2019, 24, 3080. [Google Scholar] [CrossRef]
- Yang, X.; Tong, J.; Guo, L.; Qian, Z.; Chen, Q.; Qi, R.; Qiu, Y. Bundling Potent Natural Toxin Cantharidin within Platinum (IV) Prodrugs for Liposome Drug Delivery and Effective Malignant Neuroblastoma Treatment. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 287–296. [Google Scholar] [CrossRef]
- Zhang, X.; Lin, C.; Chan, W.; Liu, K.; Lu, A.; Lin, G.; Hu, R.; Shi, H.; Zhang, H.; Yang, Z. Dual-Functional Liposomes with Carbonic Anhydrase IX Antibody and BR2 Peptide Modification Effectively Improve Intracellular Delivery of Cantharidin to Treat Orthotopic Hepatocellular Carcinoma Mice. Molecules 2019, 24, 3332. [Google Scholar] [CrossRef]
- Sun, S.; Shang, E.; Ju, A.; Li, Y.; Wu, Q.; Li, Q.; Yang, Y.; Guo, Y.; Yang, D.; Lv, S. Tumor-Targeted Hyaluronic Acid-MPEG Modified Nanostructured Lipid Carriers for Cantharidin Delivery: An in Vivo and in Vitro Study. Fitoterapia 2021, 155, 105033. [Google Scholar] [CrossRef] [PubMed]
- Braga, C.B.; Pilli, R.A.; Ornelas, C.; Weck, M. Near-Infrared Fluorescent Micelles from Poly(Norbornene) Brush Triblock Copolymers for Nanotheranostics. Biomacromolecules 2021, 22, 5290–5306. [Google Scholar] [CrossRef] [PubMed]
- Patra, D.; Mukherjee, S.; Chakraborty, I.; Dash, T.K.; Senapati, S.; Bhattacharyya, R.; Shunmugam, R. Iron(III) Coordinated Polymeric Nanomaterial: A Next-Generation Theranostic Agent for High-Resolution T 1 -Weighted Magnetic Resonance Imaging and Anticancer Drug Delivery. ACS Biomater. Sci. Eng. 2018, 4, 1738–1749. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
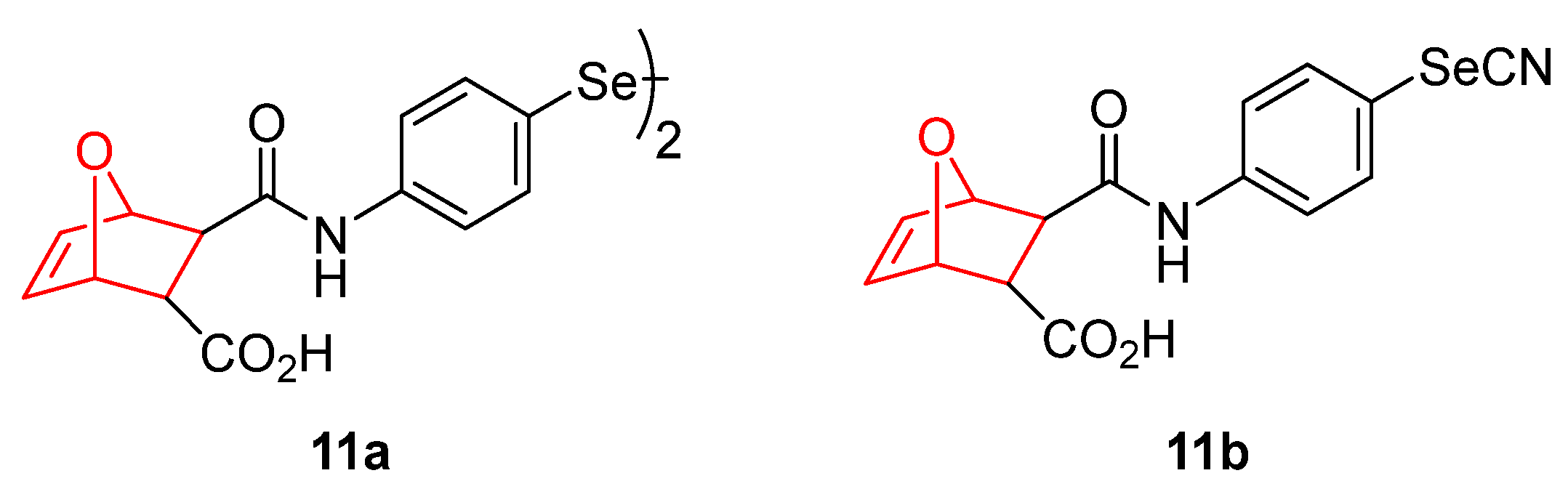{kind=link}
| MDA-MB-453 Ki, nM | MDA-MB-453 IC50, nM | LNCaP Ki, nM | LNCaP IC50, nM | PCa2b IC50, nM | |
|---|---|---|---|---|---|
| Bicalutamide | 64 | 173 | 35 | 400 | 725 |
| 2a | 158 | 307 | 6 | 41 | 180 |
| 2b | 1 | >5 | 0.5 | Ag * | Ag * |
| 2c | 5 | 12 | 2 | Ag * | Ag * |
| 2g | 2 | 30 | ND | ND | ND |
| IC50 (µM) | |||
|---|---|---|---|
| HCT-116 | HepG2 | A549 | |
| 8c | 4.45 ± 0.33 [83] | 10.74 ± 1.05 [83] | 7.72 ± 0.64 [83] |
| 8d | 9.69 ± 0.91 [83] | 7.86 ± 0.61 [83] | 46.43 ± 3.86 [83] |
| 8e | 9.14 ± 0.27 [82] | 16.70 ± 0.48 [82] | 57.82 ± 2.17 [82] |
| Cisplatin | 6.50 ± 0.47 | 3.65 ± 0.31 | 3.86 ± 0.28 |
| Carboplatin | 44.53 ± 4.64 | 39.08 ± 2.95 | 55.20 ± 4.76 |
| Oxaliplatin | 3.28 ± 0.34 | 12.80 ± 1.28 | 8.95 ± 0.78 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo-Martín, G.; Plano, D.; Martínez-Sáez, N.; Aydillo, C.; Moreno, E.; Espuelas, S.; Sanmartín, C. Norbornene and Related Structures as Scaffolds in the Search for New Cancer Treatments. Pharmaceuticals 2022, 15, 1465. https://doi.org/10.3390/ph15121465
Calvo-Martín G, Plano D, Martínez-Sáez N, Aydillo C, Moreno E, Espuelas S, Sanmartín C. Norbornene and Related Structures as Scaffolds in the Search for New Cancer Treatments. Pharmaceuticals. 2022; 15(12):1465. https://doi.org/10.3390/ph15121465
Chicago/Turabian StyleCalvo-Martín, Gorka, Daniel Plano, Nuria Martínez-Sáez, Carlos Aydillo, Esther Moreno, Socorro Espuelas, and Carmen Sanmartín. 2022. "Norbornene and Related Structures as Scaffolds in the Search for New Cancer Treatments" Pharmaceuticals 15, no. 12: 1465. https://doi.org/10.3390/ph15121465
APA StyleCalvo-Martín, G., Plano, D., Martínez-Sáez, N., Aydillo, C., Moreno, E., Espuelas, S., & Sanmartín, C. (2022). Norbornene and Related Structures as Scaffolds in the Search for New Cancer Treatments. Pharmaceuticals, 15(12), 1465. https://doi.org/10.3390/ph15121465