
Utilization of Cancer Cell Line Screening to Elucidate the Anticancer Activity and Biological Pathways Related to the Ruthenium-Based Therapeutic BOLD-100

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cancer Cell Screen
2.2. Calculation of BOLD-100 and Cisplatin Sensitivity Profile in Cancer Cell Lines
2.3. Comparison of BOLD-100 and Cisplatin Sensitivity Profiles versus GDSC Database
2.4. Identification of BOLD-100 Response Associated Genes
2.5. Functional Enrichment Analysis of BOLD-100 Related Genes
2.6. Development of Predictive Learning Model Using Gene Expression
3. Results
3.1. BOLD-100 Response Shows Variability across Cancer Cell Lines’ Tissue of Origin
3.2. BOLD-100 Exhibits Differential Cytotoxic Effects across Cancer Indications
3.3. BOLD-100 Response Shows Variability across Cell Culture Media
3.4. BOLD-100 Response Profile Shows Weak Correlation to Other Known Drugs
3.5. Multiple Regression Analysis Reveals Genes Associated to BOLD-100 Response
3.6. Pathway Enrichment Analysis of Associated Genes Reveals Key Biological Pathways
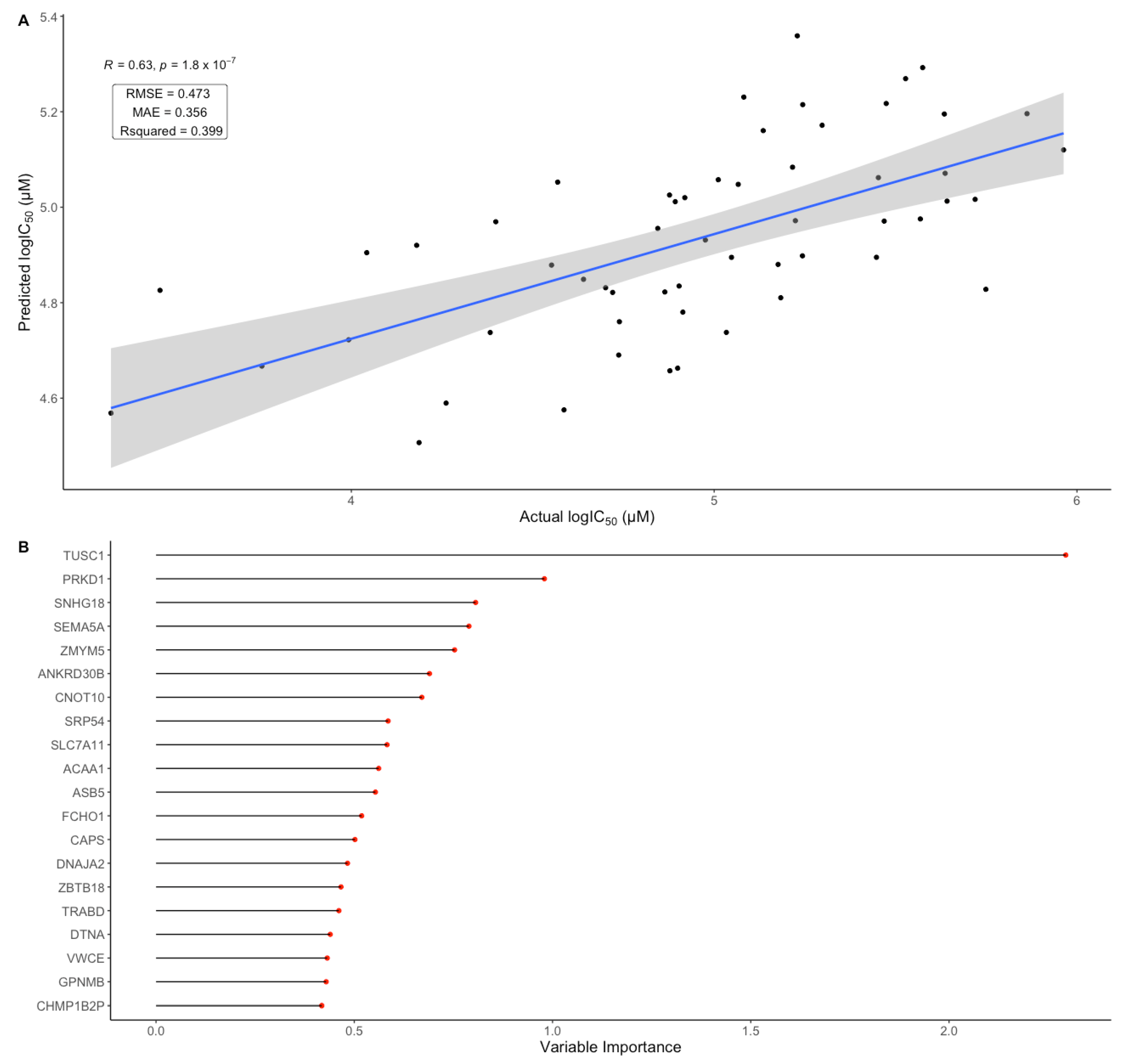
3.7. Cell Line Gene Expression Data Shows Predictive Potential for BOLD-100 Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frezza, M.; Hindo, S.; Chen, D.; Davenport, A.; Schmitt, S.; Tomco, D.; Dou, Q.P. Novel Metals and Metal Complexes as Platforms for Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal Complexes in Cancer Therapy—An Update from Drug Design Perspective. Drug Des. Devel. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.K.; Melchart, M.; Habtemariam, A.; Sadler, P.J. Organometallic Chemistry, Biology and Medicine: Ruthenium Arene Anticancer Complexes. Chem. Commun. 2005, 38, 4764–4776. [Google Scholar] [CrossRef] [PubMed]
- Haas, K.L.; Franz, K.J. Application of Metal Coordination Chemistry to Explore and Manipulate Cell Biology. Chem. Rev. 2009, 109, 4921–4960. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef]
- Desoize, B.; Madoulet, C. Particular Aspects of Platinum Compounds Used at Present in Cancer Treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 317–325. [Google Scholar] [CrossRef]
- Amable, L. Cisplatin Resistance and Opportunities for Precision Medicine. Pharmacol. Res. 2016, 106, 27–36. [Google Scholar] [CrossRef]
- Wernitznig, D.; Kiakos, K.; Del Favero, G.; Harrer, N.; Machat, H.; Osswald, A.; Jakupec, M.A.; Wernitznig, A.; Sommergruber, W.; Keppler, B.K. First-in-Class Ruthenium Anticancer Drug (KP1339/IT-139) Induces an Immunogenic Cell Death Signature in Colorectal Spheroids in Vitro†. Metallomics 2019, 11, 1044–1048. [Google Scholar] [CrossRef]
- Konda, P.; Lifshits, L.M.; Roque, J.A.; Cole, H.D.; Cameron, C.G.; McFarland, S.A.; Gujar, S. Discovery of Immunogenic Cell Death-Inducing Ruthenium-Based Photosensitizers for Anticancer Photodynamic Therapy. OncoImmunology 2021, 10, 1863626. [Google Scholar] [CrossRef] [PubMed]
- Le, H.V.; Babak, M.V.; Ehsan, M.A.; Altaf, M.; Reichert, L.; Gushchin, A.L.; Ang, W.H.; Isab, A.A. Highly Cytotoxic Gold(I)-Phosphane Dithiocarbamate Complexes Trigger an ER Stress-Dependent Immune Response in Ovarian Cancer Cells. Dalton Trans. 2020, 49, 7355–7363. [Google Scholar] [CrossRef]
- Xiong, X.; Huang, K.-B.; Wang, Y.; Cao, B.; Luo, Y.; Chen, H.; Yang, Y.; Long, Y.; Liu, M.; Chan, A.S.C.; et al. Target Profiling of an Iridium(III)-Based Immunogenic Cell Death Inducer Unveils the Engagement of Unfolded Protein Response Regulator BiP. J. Am. Chem. Soc. 2022, 144, 10407–10416. [Google Scholar] [CrossRef] [PubMed]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M.; et al. Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Kar, B.; Pete, S.; Paira, P. Ru(II), Ir(III), Re(I) and Rh(III) Based Complexes as next Generation Anticancer Metallopharmaceuticals. Dalton Trans. 2021, 50, 11259–11290. [Google Scholar] [CrossRef]
- Wilding, J.L.; Bodmer, W.F. Cancer Cell Lines for Drug Discovery and Development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging Landscape of Oncogenic Signatures across Human Cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Tipney, H.; Painter, J.L.; Shen, J.; Nicoletti, P.; Shen, Y.; Floratos, A.; Sham, P.C.; Li, M.J.; Wang, J.; et al. The Support of Human Genetic Evidence for Approved Drug Indications. Nat. Genet. 2015, 47, 856–860. [Google Scholar] [CrossRef]
- Adam, G.; Rampášek, L.; Safikhani, Z.; Smirnov, P.; Haibe-Kains, B.; Goldenberg, A. Machine Learning Approaches to Drug Response Prediction: Challenges and Recent Progress. Npj Precis. Oncol. 2020, 4, 19. [Google Scholar] [CrossRef]
- Xia, F.; Allen, J.; Balaprakash, P.; Brettin, T.; Garcia-Cardona, C.; Clyde, A.; Cohn, J.; Doroshow, J.; Duan, X.; Dubinkina, V.; et al. A Cross-Study Analysis of Drug Response Prediction in Cancer Cell Lines. Brief. Bioinform. 2022, 23, bbab356. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating Chemical Sensitivity and Basal Gene Expression Reveals Mechanism of Action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2013, 41, D955-61. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-Generation Characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Haverty, P.M.; Lin, E.; Tan, J.; Yu, Y.; Lam, B.; Lianoglou, S.; Neve, R.M.; Martin, S.; Settleman, J.; Yauch, R.L.; et al. Reproducible Pharmacogenomic Profiling of Cancer Cell Line Panels. Nature 2016, 533, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Kurilov, R.; Haibe-Kains, B.; Brors, B. Assessment of Modelling Strategies for Drug Response Prediction in Cell Lines and Xenografts. Sci. Rep. 2020, 10, 2849. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Zhang, N.; Li, C.; Wang, H.; Fang, Y.; Wang, J.; Zheng, X. Anticancer Drug Sensitivity Prediction in Cell Lines from Baseline Gene Expression through Recursive Feature Selection. BMC Cancer 2015, 15, 489. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Aittokallio, T. Machine Learning and Feature Selection for Drug Response Prediction in Precision Oncology Applications. Biophys. Rev. 2018, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Menden, M.P.; Iorio, F.; Garnett, M.; McDermott, U.; Benes, C.H.; Ballester, P.J.; Saez-Rodriguez, J. Machine Learning Prediction of Cancer Cell Sensitivity to Drugs Based on Genomic and Chemical Properties. PLoS ONE 2013, 8, e61318. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modeling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Papillon-Cavanagh, S.; De Jay, N.; Hachem, N.; Olsen, C.; Bontempi, G.; Aerts, H.J.W.L.; Quackenbush, J.; Haibe-Kains, B. Comparison and Validation of Genomic Predictors for Anticancer Drug Sensitivity. J. Am. Med. Inform. Assoc. JAMIA 2013, 20, 597–602. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of Clinical Drug Development Fails and How to Improve It? Acta Pharm. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Burris, H.A.; Bakewell, S.; Bendell, J.C.; Infante, J.; Jones, S.F.; Spigel, D.R.; Weiss, G.J.; Ramanathan, R.K.; Ogden, A.; Von Hoff, D. Safety and Activity of IT-139, a Ruthenium-Based Compound, in Patients with Advanced Solid Tumours: A First-in-Human, Open-Label, Dose-Escalation Phase I Study with Expansion Cohort. ESMO Open 2017, 1, e000154. [Google Scholar] [CrossRef]
- Bakewell, S.J.; Rangel, D.F.; Ha, D.P.; Sethuraman, J.; Crouse, R.; Hadley, E.; Costich, T.L.; Zhou, X.; Nichols, P.; Lee, A.S. Suppression of Stress Induction of the 78-Kilodalton Glucose Regulated Protein (GRP78) in Cancer by IT-139, an Anti-Tumor Ruthenium Small Molecule Inhibitor. Oncotarget 2018, 9, 29698–29714. [Google Scholar] [CrossRef] [PubMed]
- Bakewell, S.; Conde, I.; Fallah, Y.; McCoy, M.; Jin, L.; Shajahan-Haq, A.N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers 2020, 12, 2647. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Atil, B.; Kryeziu, K.; Groza, D.; Koellensperger, G.; Körner, W.; Jungwirth, U.; Mohr, T.; Keppler, B.K.; Berger, W. The Ruthenium Compound KP1339 Potentiates the Anticancer Activity of Sorafenib in Vitro and in Vivo. Eur. J. Cancer Oxf. Engl. 1990 2013, 49, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Kapitza, S.; Jakupec, M.A.; Uhl, M.; Keppler, B.K.; Marian, B. The Heterocyclic Ruthenium(III) Complex KP1019 (FFC14A) Causes DNA Damage and Oxidative Stress in Colorectal Tumor Cells. Cancer Lett. 2005, 226, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Flocke, L.S.; Trondl, R.; Jakupec, M.A.; Keppler, B.K. Molecular Mode of Action of NKP-1339—A Clinically Investigated Ruthenium-Based Drug—Involves ER- and ROS-Related Effects in Colon Carcinoma Cell Lines. Investig. New Drugs 2016, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Carson, R.; Karelia, S.; Lavin, D.; Tiwari, V.; Kennedy, R.; Savage, K.; Carie, A.; Pankovich, J.; Bazett, M.; Van Schaeybroeck, S. Abstract 1183: Targeting the DNA Repair Pathway with BOLD-100 in BRAF Mutant Colorectal Cancer. Cancer Res. 2021, 81, 1183. [Google Scholar] [CrossRef]
- Schoenhacker-Alte, B.; Mohr, T.; Pirker, C.; Kryeziu, K.; Kuhn, P.-S.; Buck, A.; Hofmann, T.; Gerner, C.; Hermann, G.; Koellensperger, G.; et al. Sensitivity towards the GRP78 Inhibitor KP1339/IT-139 Is Characterized by Apoptosis Induction via Caspase 8 upon Disruption of ER Homeostasis. Cancer Lett. 2017, 404, 79–88. [Google Scholar] [CrossRef]
- Neuditschko, B.; Legin, A.A.; Baier, D.; Schintlmeister, A.; Reipert, S.; Wagner, M.; Keppler, B.K.; Berger, W.; Meier-Menches, S.M.; Gerner, C. Interaction with Ribosomal Proteins Accompanies Stress Induction of the Anticancer Metallodrug BOLD-100/KP1339 in the Endoplasmic Reticulum. Angew. Chem. Int. Ed. 2021, 60, 5063–5068. [Google Scholar] [CrossRef]
- Rusz, M.; Rampler, E.; Keppler, B.K.; Jakupec, M.A.; Koellensperger, G. Single Spheroid Metabolomics: Optimizing Sample Preparation of Three-Dimensional Multicellular Tumor Spheroids. Metabolites 2019, 9, 304. [Google Scholar] [CrossRef]
- Baier, D.; Schoenhacker-Alte, B.; Rusz, M.; Pirker, C.; Mohr, T.; Mendrina, T.; Kirchhofer, D.; Meier-Menches, S.M.; Hohenwallner, K.; Schaier, M.; et al. The Anticancer Ruthenium Compound BOLD-100 Targets Glycolysis and Generates a Metabolic Vulnerability towards Glucose Deprivation. Pharmaceutics 2022, 14, 238. [Google Scholar] [CrossRef]
- Herrmann, H.A.; Rusz, M.; Baier, D.; Jakupec, M.A.; Keppler, B.K.; Berger, W.; Koellensperger, G.; Zanghellini, J. Thermodynamic Genome-Scale Metabolic Modeling of Metallodrug Resistance in Colorectal Cancer. Cancers 2021, 13, 4130. [Google Scholar] [CrossRef] [PubMed]
- Smirnov, P.; Safikhani, Z.; El-Hachem, N.; Wang, D.; She, A.; Olsen, C.; Freeman, M.; Selby, H.; Gendoo, D.M.A.; Grossmann, P.; et al. PharmacoGx: An R Package for Analysis of Large Pharmacogenomic Datasets. Bioinformatics 2016, 32, 1244–1246. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.A.; Hafner, M.; Kouril, M.; Williams, E.H.; Muhlich, J.L.; Pilarczyk, M.; Niepel, M.; Sorger, P.K.; Medvedovic, M. GRcalculator: An Online Tool for Calculating and Mining Dose–Response Data. BMC Cancer 2017, 17, 698. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, D.; Barthorpe, S.; Yang, W.; Lightfoot, H.; Hall, C.; Gilbert, J.; Francies, H.E.; Garnett, M.J. Cell Model Passports—A Hub for Clinical, Genetic and Functional Datasets of Preclinical Cancer Models. Nucleic Acids Res. 2019, 47, D923–D929. [Google Scholar] [CrossRef]
- Zuiverloon, T.C.M.; de Jong, F.C.; Costello, J.C.; Theodorescu, D. Systematic Review: Characteristics and Preclinical Uses of Bladder Cancer Cell Lines. Bladder Cancer 2018, 4, 169–183. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Huss, W.; Foster, B.; Ohm, J.; Wang, J.; Azabdaftari, G.; Eng, K.H.; Woloszynska-Read, A. Transcriptional Changes Associated with in Vivo Growth of Muscle-Invasive Bladder Cancer Cell Lines in Nude Mice. Am. J. Clin. Exp. Urol. 2018, 6, 138–148. [Google Scholar]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The Molecular Landscape of Colorectal Cancer Cell Lines Unveils Clinically Actionable Kinase Targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef]
- Hirschfield, H.; Bian, C.B.; Higashi, T.; Nakagawa, S.; Zeleke, T.Z.; Nair, V.D.; Fuchs, B.C.; Hoshida, Y. In Vitro Modeling of Hepatocellular Carcinoma Molecular Subtypes for Anti-Cancer Drug Assessment. Exp. Mol. Med. 2018, 50, e419. [Google Scholar] [CrossRef]
- Iorio, F.; Knijnenburg, T.A.; Vis, D.J.; Bignell, G.R.; Menden, M.P.; Schubert, M.; Aben, N.; Gonçalves, E.; Barthorpe, S.; Lightfoot, H.; et al. A Landscape of Pharmacogenomic Interactions in Cancer. Cell 2016, 166, 740–754. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building Predictive Models in R Using the Caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Parvandeh, S.; Yeh, H.-W.; Paulus, M.P.; McKinney, B.A. Consensus Features Nested Cross-Validation. Bioinformatics 2020, 36, 3093–3098. [Google Scholar] [CrossRef]
- Yang, X.; Wen, Y.; Song, X.; He, S.; Bo, X. Exploring the Classification of Cancer Cell Lines from Multiple Omic Views. PeerJ 2020, 8, e9440. [Google Scholar] [CrossRef]
- Kummar, S.; Gutierrez, M.; Doroshow, J.H.; Murgo, A.J. Drug Development in Oncology: Classical Cytotoxics and Molecularly Targeted Agents. Br. J. Clin. Pharmacol. 2006, 62, 15–26. [Google Scholar] [CrossRef]
- Rixe, O.; Fojo, T. Is Cell Death a Critical End Point for Anticancer Therapies or Is Cytostasis Sufficient? Clin. Cancer Res. 2007, 13, 7280–7287. [Google Scholar] [CrossRef]
- Anttila, J.V.; Shubin, M.; Cairns, J.; Borse, F.; Guo, Q.; Mononen, T.; Vázquez-García, I.; Pulkkinen, O.; Mustonen, V. Contrasting the Impact of Cytotoxic and Cytostatic Drug Therapies on Tumour Progression. PLoS Comput. Biol. 2019, 15, e1007493. [Google Scholar] [CrossRef]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth Rate Inhibition Metrics Correct for Confounders in Measuring Sensitivity to Cancer Drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef]
- Larsson, P.; Engqvist, H.; Biermann, J.; Werner Rönnerman, E.; Forssell-Aronsson, E.; Kovács, A.; Karlsson, P.; Helou, K.; Parris, T.Z. Optimization of Cell Viability Assays to Improve Replicability and Reproducibility of Cancer Drug Sensitivity Screens. Sci. Rep. 2020, 10, 5798. [Google Scholar] [CrossRef]
- Niepel, M.; Hafner, M.; Mills, C.E.; Subramanian, K.; Williams, E.H.; Chung, M.; Gaudio, B.; Barrette, A.M.; Stern, A.D.; Hu, B.; et al. A Multi-Center Study on the Reproducibility of Drug-Response Assays in Mammalian Cell Lines. Cell Syst. 2019, 9, 35–48.e5. [Google Scholar] [CrossRef] [PubMed]
- Naasani, I. COMPARE Analysis, a Bioinformatic Approach to Accelerate Drug Repurposing against Covid-19 and Other Emerging Epidemics. SLAS Discov. Adv. Sci. Drug Discov. 2021, 26, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Zaharevitz, D.W.; Holbeck, S.L.; Bowerman, C.; Svetlik, P.A. COMPARE: A Web Accessible Tool for Investigating Mechanisms of Cell Growth Inhibition. J. Mol. Graph. Model. 2002, 20, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Krushkal, J.; Negi, S.; Yee, L.M.; Evans, J.R.; Grkovic, T.; Palmisano, A.; Fang, J.; Sankaran, H.; McShane, L.M.; Zhao, Y.; et al. Molecular Genomic Features Associated with in Vitro Response of the NCI-60 Cancer Cell Line Panel to Natural Products. Mol. Oncol. 2021, 15, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tchounwou, P.B. Molecular Mechanisms of Cisplatin Cytotoxicity in Acute Promyelocytic Leukemia Cells. Oncotarget 2015, 6, 40734–40746. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Mahadevan, D.; Plummer, R.; Squires, M.S.; Rensvold, D.; Kurtin, S.; Pretzinger, C.; Dragovich, T.; Adams, J.; Lock, V.; Smith, D.M.; et al. A Phase I Pharmacokinetic and Pharmacodynamic Study of AT7519, a Cyclin-Dependent Kinase Inhibitor in Patients with Refractory Solid Tumors. Ann. Oncol. 2011, 22, 2137–2143. [Google Scholar] [CrossRef]
- Squires, M.S.; Feltell, R.E.; Wallis, N.G.; Lewis, E.J.; Smith, D.-M.; Cross, D.M.; Lyons, J.F.; Thompson, N.T. Biological Characterization of AT7519, a Small-Molecule Inhibitor of Cyclin-Dependent Kinases, in Human Tumor Cell Lines. Mol. Cancer Ther. 2009, 8, 324–332. [Google Scholar] [CrossRef]
- Koras, K.; Juraeva, D.; Kreis, J.; Mazur, J.; Staub, E.; Szczurek, E. Feature Selection Strategies for Drug Sensitivity Prediction. Sci. Rep. 2020, 10, 9377. [Google Scholar] [CrossRef] [PubMed]
- Sharifi-Noghabi, H.; Jahangiri-Tazehkand, S.; Smirnov, P.; Hon, C.; Mammoliti, A.; Nair, S.K.; Mer, A.S.; Ester, M.; Haibe-Kains, B. Drug Sensitivity Prediction from Cell Line-Based Pharmacogenomics Data: Guidelines for Developing Machine Learning Models. Brief. Bioinform. 2021, 22, bbab294. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Guo, X.; Yu, H. Variable Selection Using Mean Decrease Accuracy and Mean Decrease Gini Based on Random Forest. In Proceedings of the 2016 7th IEEE International Conference on Software Engineering and Service Science (ICSESS), Beijing, China, 26–28 August 2016; pp. 219–224. [Google Scholar]
- Köberle, B.; Schoch, S. Platinum Complexes in Colorectal Cancer and Other Solid Tumors. Cancers 2021, 13, 2073. [Google Scholar] [CrossRef] [PubMed]
- Groessl, M.; Zava, O.; Dyson, P.J. Cellular Uptake and Subcellular Distribution of Ruthenium-Based Metallodrugs under Clinical Investigation versus Cisplatin. Metallomics 2011, 3, 591. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Chang, J.-Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Shah, M.A.; Muro, K.; Francois, E.; Adenis, A.; Hsu, C.-H.; Doi, T.; Moriwaki, T.; Kim, S.-B.; Lee, S.-H.; et al. Randomized Phase III KEYNOTE-181 Study of Pembrolizumab Versus Chemotherapy in Advanced Esophageal Cancer. J. Clin. Oncol. 2020, 38, 4138–4148. [Google Scholar] [CrossRef]
- Kato, K.; Cho, B.C.; Takahashi, M.; Okada, M.; Lin, C.-Y.; Chin, K.; Kadowaki, S.; Ahn, M.-J.; Hamamoto, Y.; Doki, Y.; et al. Nivolumab versus Chemotherapy in Patients with Advanced Oesophageal Squamous Cell Carcinoma Refractory or Intolerant to Previous Chemotherapy (ATTRACTION-3): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 1506–1517. [Google Scholar] [CrossRef]
- Sun, J.-M.; Shen, L.; Shah, M.A.; Enzinger, P.; Adenis, A.; Doi, T.; Kojima, T.; Metges, J.-P.; Li, Z.; Kim, S.-B.; et al. Pembrolizumab plus Chemotherapy versus Chemotherapy Alone for First-Line Treatment of Advanced Oesophageal Cancer (KEYNOTE-590): A Randomised, Placebo-Controlled, Phase 3 Study. Lancet 2021, 398, 759–771. [Google Scholar] [CrossRef]
- Bytzek, A.K.; Koellensperger, G.; Keppler, B.K.; Hartinger, C. Biodistribution of the Novel Anticancer Drug Sodium Trans-[Tetrachloridobis(1H-Indazole)Ruthenate(III)] KP-1339/IT139 in Nude BALB/c Mice and Implications on Its Mode of Action. J. Inorg. Biochem. 2016, 160, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, C.; Ishikawa, Y.; Honjoh, D.; Kimura, M.; Shimane, M.; Miyoshi, T.; Nomura, H.; Jones, M.H. Integrated Classification of Lung Tumors and Cell Lines by Expression Profiling. Proc. Natl. Acad. Sci. USA. 2002, 99, 12357–12362. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating Cell Lines as Tumour Models by Comparison of Genomic Profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef]
- Vincent, K.M.; Findlay, S.D.; Postovit, L.M. Assessing Breast Cancer Cell Lines as Tumour Models by Comparison of MRNA Expression Profiles. Breast Cancer Res. 2015, 17, 114. [Google Scholar] [CrossRef]
- Goodspeed, A.; Heiser, L.M.; Gray, J.W.; Costello, J.C. Tumor-Derived Cell Lines as Molecular Models of Cancer Pharmacogenomics. Mol. Cancer Res. MCR 2016, 14, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, R.; Ni, M.; Gill, P.; Lee, A.S. Cell Surface Relocalization of the Endoplasmic Reticulum Chaperone and Unfolded Protein Response Regulator GRP78/BiP. J. Biol. Chem. 2010, 285, 15065–15075. [Google Scholar] [CrossRef] [PubMed]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin—From Traditional Medicine to Anticancer Drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- Gifford, J.B.; Huang, W.; Zeleniak, A.E.; Hindoyan, A.; Wu, H.; Donahue, T.R.; Hill, R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2016, 15, 1043–1052. [Google Scholar] [CrossRef]
- Burger, K.; Mühl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hölzel, M.; et al. Chemotherapeutic Drugs Inhibit Ribosome Biogenesis at Various Levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef]
- Zisi, A.; Bartek, J.; Lindström, M.S. Targeting Ribosome Biogenesis in Cancer: Lessons Learned and Way Forward. Cancers 2022, 14, 2126. [Google Scholar] [CrossRef]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP–MDM2–P53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef]
- Zhang, F.; Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Gordan, J.D.; Dai, M.-S.; Lu, H.; Simon, M.C.; Diehl, J.A. Ribosomal Stress Couples the Unfolded Protein Response to P53-Dependent Cell Cycle Arrest. J. Biol. Chem. 2006, 281, 30036–30045. [Google Scholar] [CrossRef]
- Sharma, A.; Rani, R. Ensembled Machine Learning Framework for Drug Sensitivity Prediction. IET Syst. Biol. 2020, 14, 39–46. [Google Scholar] [CrossRef]
- Riddick, G.; Song, H.; Ahn, S.; Walling, J.; Borges-Rivera, D.; Zhang, W.; Fine, H.A. Predicting in Vitro Drug Sensitivity Using Random Forests. Bioinformatics 2011, 27, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Strobl, C.; Boulesteix, A.-L.; Kneib, T.; Augustin, T.; Zeileis, A. Conditional Variable Importance for Random Forests. BMC Bioinform. 2008, 9, 307. [Google Scholar] [CrossRef] [PubMed]
- Lunetta, K.L.; Hayward, L.B.; Segal, J.; Van Eerdewegh, P. Screening Large-Scale Association Study Data: Exploiting Interactions Using Random Forests. BMC Genet. 2004, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Shan, Z.; Shakoori, A.; Bodaghi, S.; Goldsmith, P.; Jin, J.; Wiest, J.S. TUSC1, a Putative Tumor Suppressor Gene, Reduces Tumor Cell Growth In Vitro and Tumor Growth In Vivo. PLoS ONE 2013, 8, e66114. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BOLD-100 vs. GDSC—Solid Cancers | |||
|---|---|---|---|
| Drug | Pathway Name | ρ | FDR |
| AT-7519 | Cell cycle | 0.321 | 0.00290 |
| Bleomycin | DNA replication | 0.316 | 0.00290 |
| Thapsigargin | Other | 0.314 | 0.00290 |
| FMK | Other, kinases | 0.310 | 0.00327 |
| Bosutinib | Other, kinases | 0.291 | 0.00327 |
| BOLD-100 vs. GDSC—Liquid Cancers | |||
| Drug | Pathway Name | ρ | FDR |
| KRAS (G12C) Inhibitor-12 | ERK MAPK signaling | 0.658 | 0.000698 |
| ULK1_4989 | Other, kinases | 0.640 | 0.000698 |
| VSP34_8731 | Other | 0.635 | 0.000698 |
| Vincristine | Mitosis | 0.626 | 0.000698 |
| Carmustine | DNA replication | 0.622 | 0.000698 |
| Cisplatin vs. GDSC—Solid Cancers | |||
| Drug | Pathway Name | ρ | FDR |
| Cisplatin | DNA replication | 0.688 | 1.54 × 10−24 |
| Camptothecin | DNA replication | 0.547 | 6.68 × 10−14 |
| Mitoxantrone | DNA replication | 0.516 | 7.45 × 10−11 |
| Topotecan | DNA replication | 0.509 | 1.21 × 10−10 |
| Irinotecan | DNA replication | 0.473 | 5.94 × 10−10 |
| Cisplatin vs GDSC—Liquid Cancers | |||
| Drug | Pathway Name | ρ | FDR |
| Epirubicin | DNA replication | 0.792 | 1.41 × 10−8 |
| Talazoparib | Genome integrity | 0.776 | 1.48 × 10−9 |
| PARP_9482 | Genome integrity | 0.756 | 1.41 × 10−8 |
| PARP_0108 | Genome integrity | 0.747 | 1.46 × 10−8 |
| Camptothecin | DNA replication | 0.729 | 3.74 × 10−7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, B.J.; Raha, P.; Pankovich, J.; Bazett, M. Utilization of Cancer Cell Line Screening to Elucidate the Anticancer Activity and Biological Pathways Related to the Ruthenium-Based Therapeutic BOLD-100. Cancers 2023, 15, 28. https://doi.org/10.3390/cancers15010028
Park BJ, Raha P, Pankovich J, Bazett M. Utilization of Cancer Cell Line Screening to Elucidate the Anticancer Activity and Biological Pathways Related to the Ruthenium-Based Therapeutic BOLD-100. Cancers. 2023; 15(1):28. https://doi.org/10.3390/cancers15010028
Chicago/Turabian StylePark, Brian J., Paromita Raha, Jim Pankovich, and Mark Bazett. 2023. "Utilization of Cancer Cell Line Screening to Elucidate the Anticancer Activity and Biological Pathways Related to the Ruthenium-Based Therapeutic BOLD-100" Cancers 15, no. 1: 28. https://doi.org/10.3390/cancers15010028
APA StylePark, B. J., Raha, P., Pankovich, J., & Bazett, M. (2023). Utilization of Cancer Cell Line Screening to Elucidate the Anticancer Activity and Biological Pathways Related to the Ruthenium-Based Therapeutic BOLD-100. Cancers, 15(1), 28. https://doi.org/10.3390/cancers15010028