







Pharmaceuticals 2026, 19(7), 1035; https://doi.org/10.3390/ph19071035 - 2 Jul 2026
Abstract
Background: Intensive low-density lipoprotein cholesterol (LDL-C) reduction is essential for secondary prevention after acute coronary syndrome (ACS). Although randomized trials support the use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, real-world evidence on long-term lipid control and the optimal timing of
[...] Read more.
Background: Intensive low-density lipoprotein cholesterol (LDL-C) reduction is essential for secondary prevention after acute coronary syndrome (ACS). Although randomized trials support the use of proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors, real-world evidence on long-term lipid control and the optimal timing of initiation remains limited. Objective: To evaluate lipid outcomes and atherosclerotic cardiovascular disease (ASCVD) events associated with evolocumab after ACS and to assess whether timing of initiation influences these outcomes in routine clinical practice. Methods: This multicenter, retrospective cohort study included adults who were initiated on evolocumab following ACS between 2017 and 2024. Lipid parameters were assessed at predefined follow-up time points (3 months, 6 months, 1 year, 2 years, and up to 3 years) using an available-case approach. Patients were categorized as early initiators (≤1 month) or late initiators (>1 month). Recurrent ASCVD events, hospitalization burden, and mortality were analyzed using multivariable regression. A propensity score-matched sensitivity analysis was also performed. Results: Among 525 included patients (mean age 53.1 ± 11.6 years; 80.2% male), baseline LDL-C was 3.68 ± 1.66 mmol/L. LDL-C decreased to approximately 1.8–2.0 mmol/L within 3 months, corresponding to an approximate 45–50% reduction from baseline, with consistent reductions observed across available follow-up time points. Recurrent ASCVD events occurred in 14.8% of patients, and in-hospital mortality was 2.3%. Although early initiators had higher baseline risk, adjusted analyses showed no statistically significant association between early initiation and recurrent ASCVD (adjusted OR 1.50; 95% CI 0.82–2.70; p = 0.17). Similarly, no statistically significant differences in lipid outcomes were observed between early and late initiation groups after adjustment. Findings were consistent in propensity score-matched analyses. Conclusions: In this real-world post-ACS cohort, evolocumab was associated with substantial and sustained LDL-C reduction across follow-up time points. No significant associations were observed between timing of initiation and lipid or ASCVD outcomes after adjustment. These findings should be interpreted cautiously, given the observational design, available-case analysis, and potential for residual confounding.
Full article
(This article belongs to the Special Issue Advancements in Cardiovascular and Antidiabetic Drug Therapy, 2nd Edition)
►
Show Figures
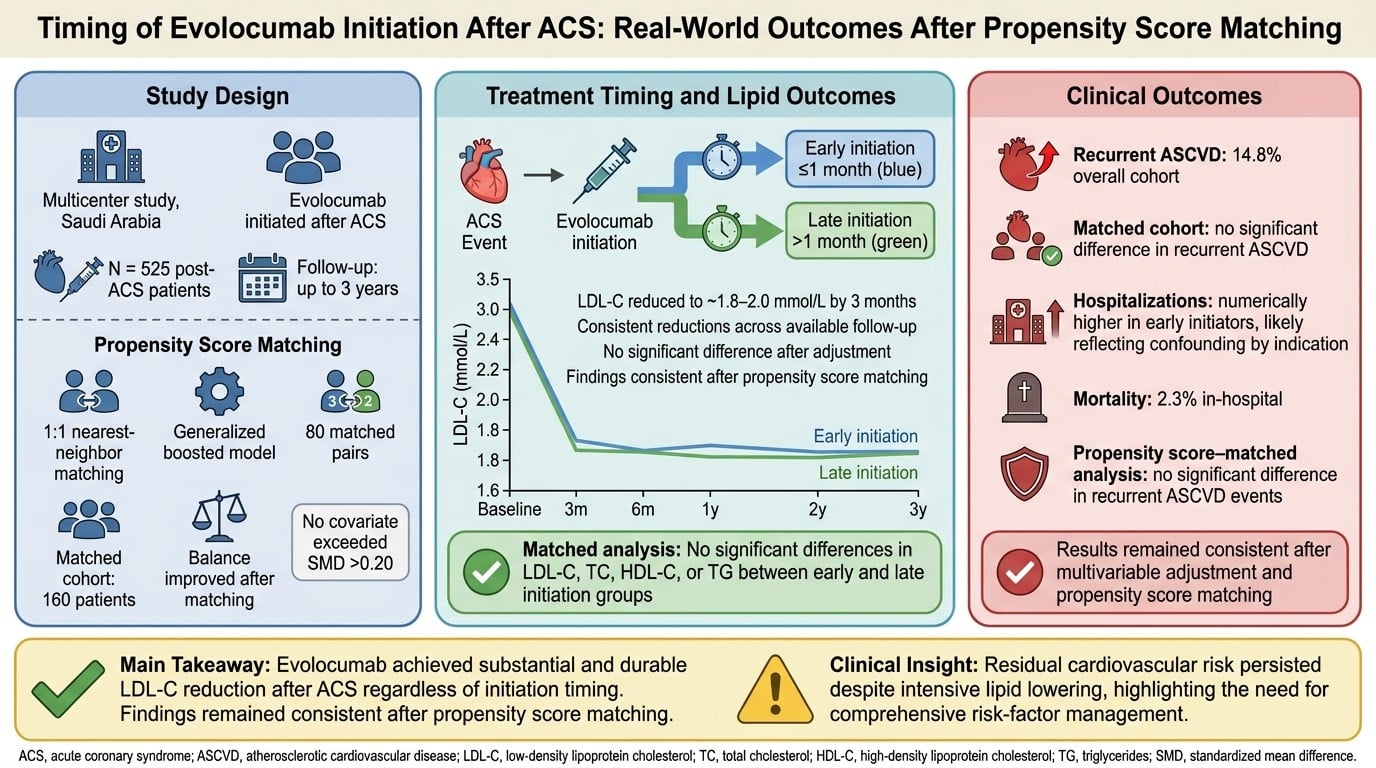
Graphical abstract






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}