
TRAIL Receptor Targeting Agents Potentiate PARP Inhibitor Efficacy in Pancreatic Cancer Independently of BRCA2 Mutation Status

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Patient-Derived Organoid Lines
2.3. Cell Proliferation Assay
2.4. Clonogenic Assay
2.5. Organoid CellTiter-Glo 3D Viability Assay
2.6. Western Blot
2.7. Flow Cytometry of Surface Receptors
2.8. Cell Cycle and Apoptosis Assays
2.9. Immunofluorescence Microscopy
2.10. Statistics
3. Results
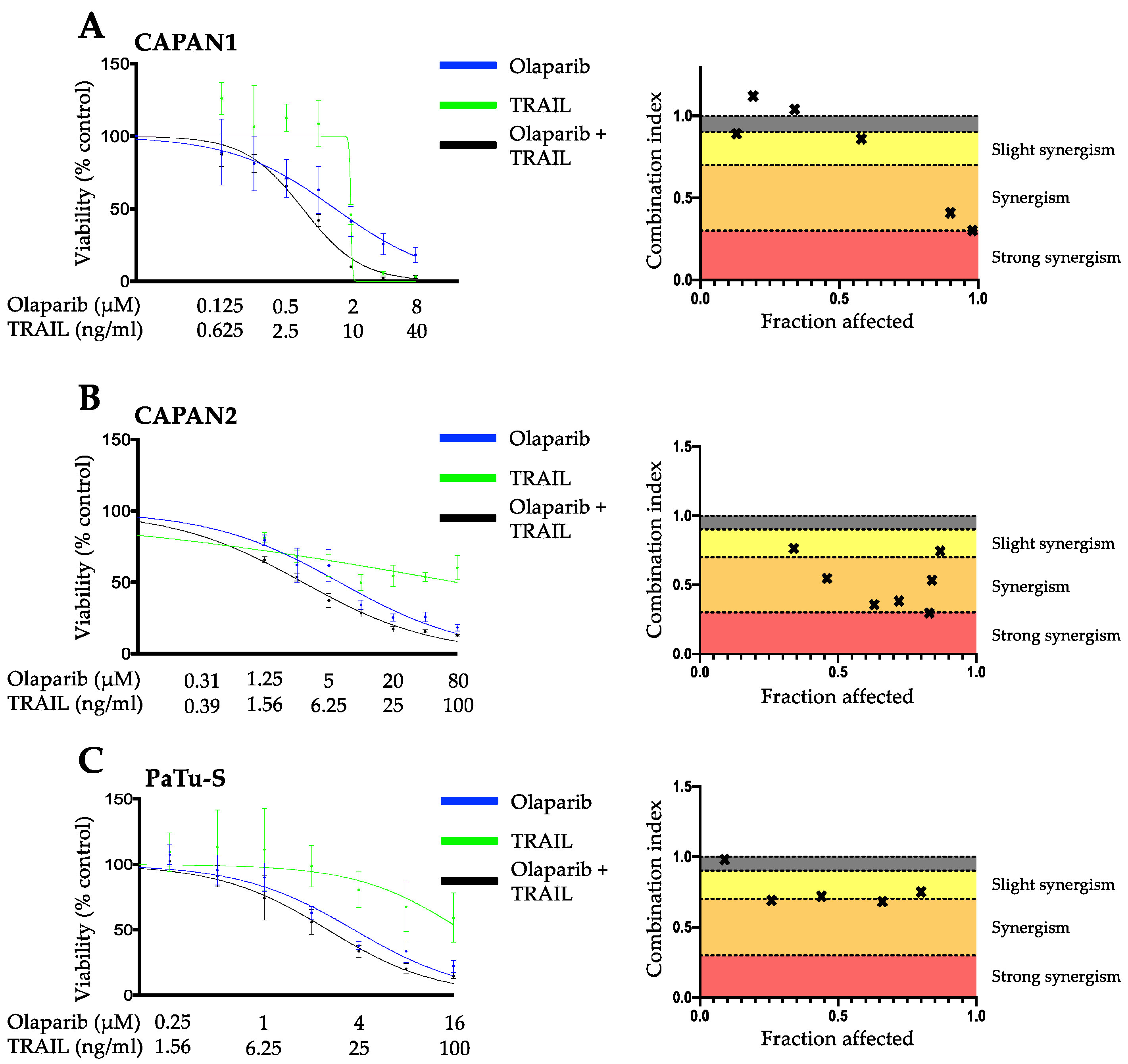
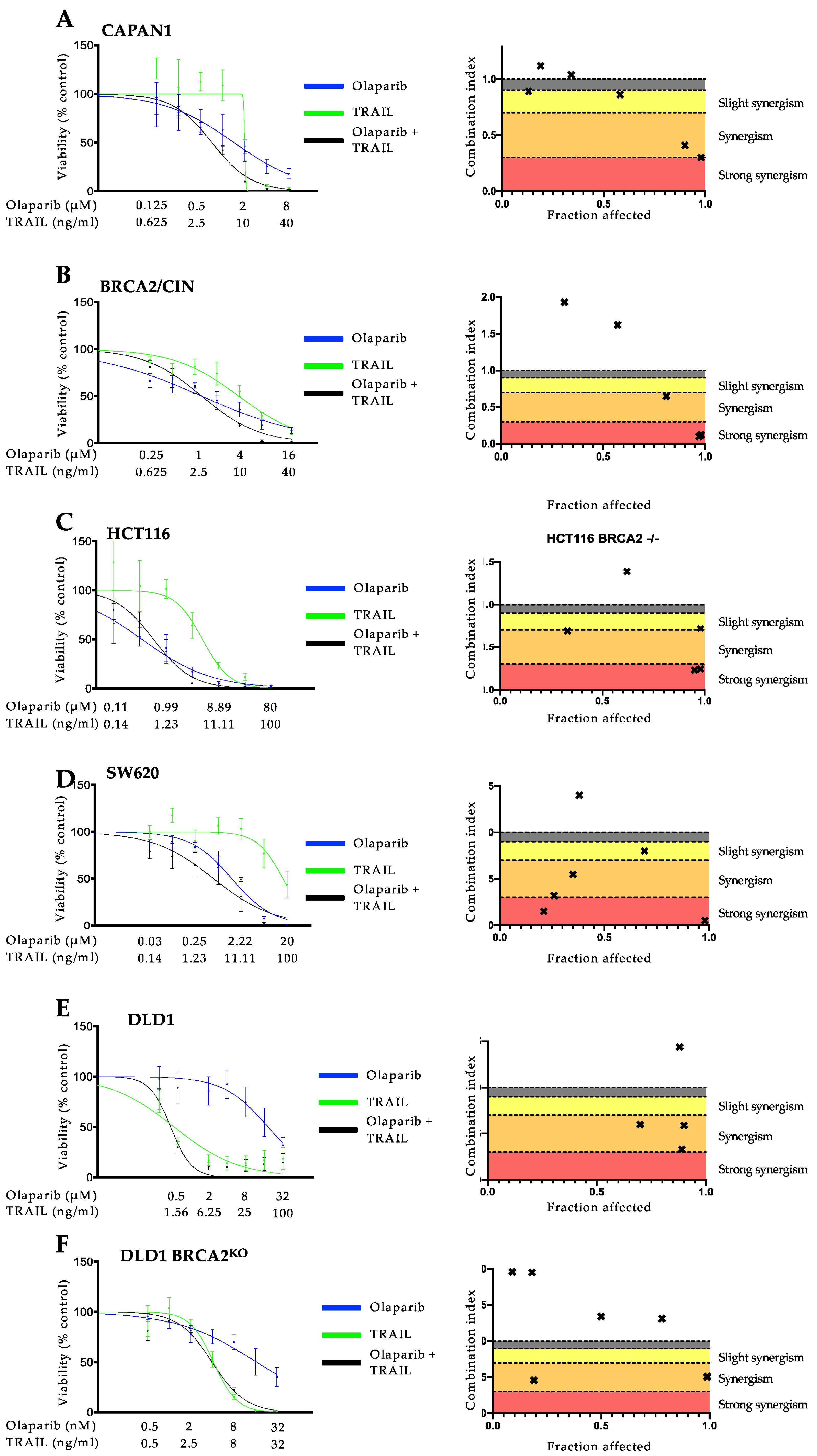
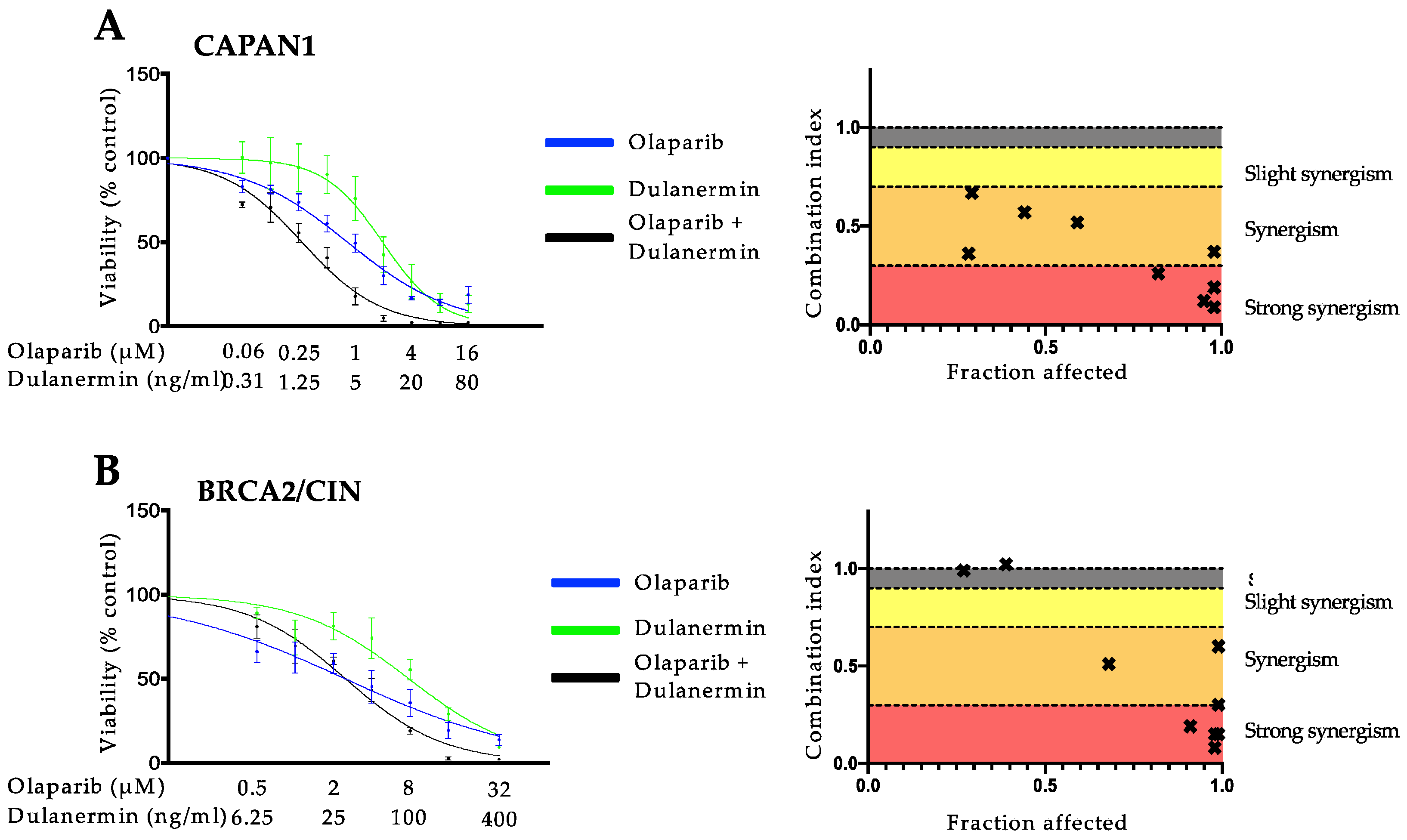
3.1. PARP Inhibition and TRAIL Synergize to Cause Loss of Cell Viability in Pancreatic Cancer Cell Lines
3.2. The Synergistic Interaction between Olaparib and TRAIL Is Independent of BRCA2 Mutational Status
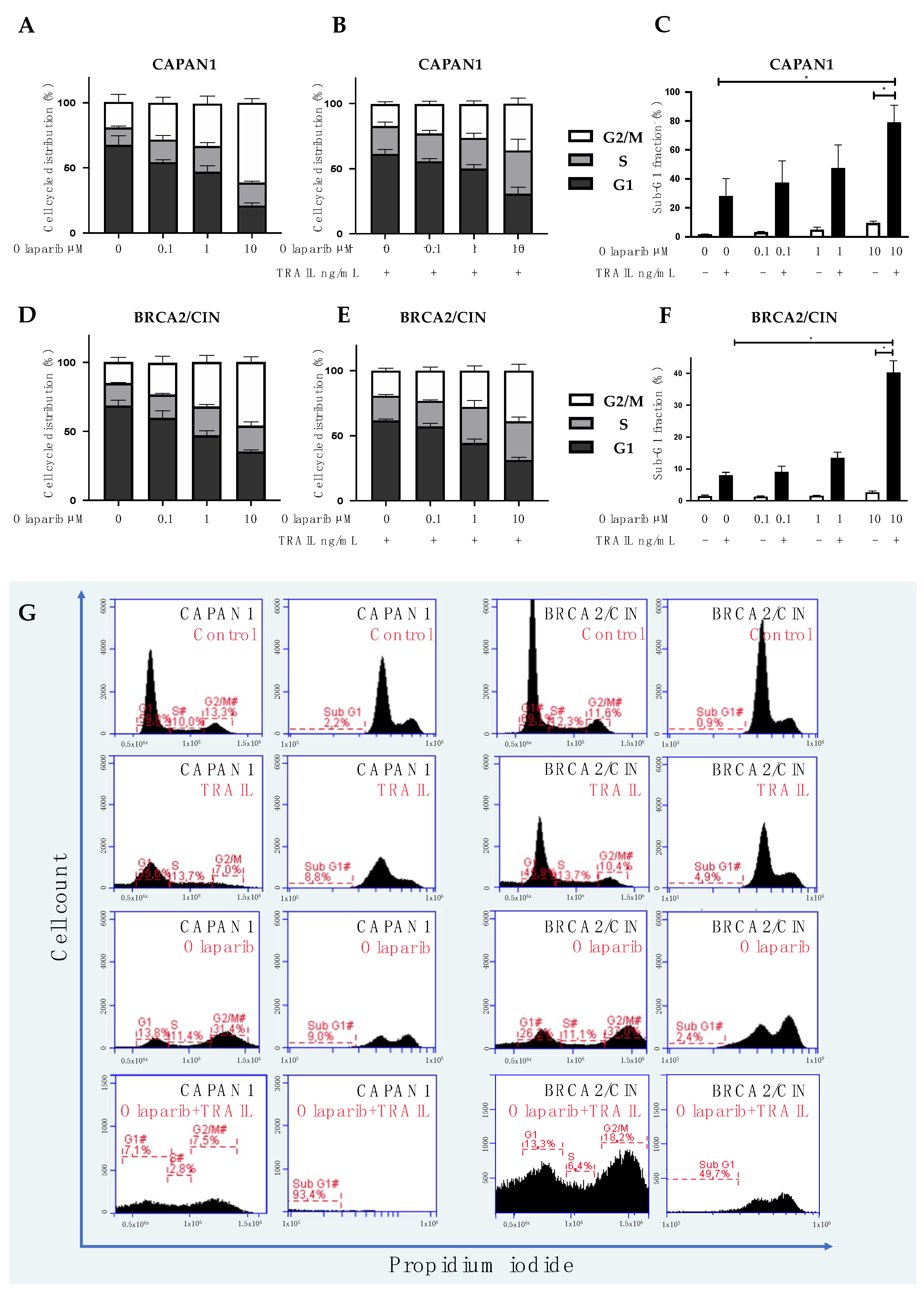
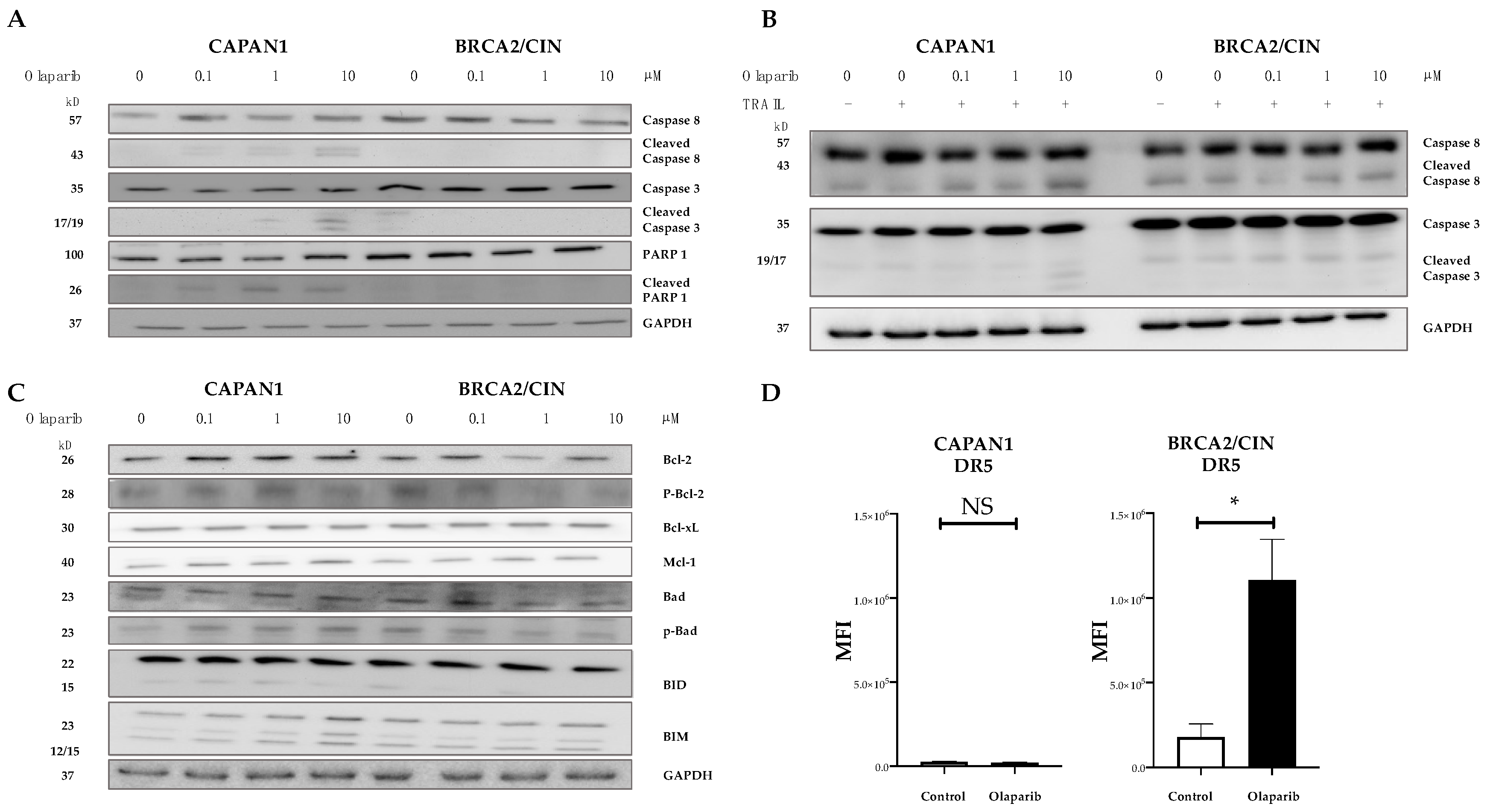
3.3. Combined Olaparib and TRAIL Enhance S/G2 Phase Cell Cycle Arrest and Apoptosis
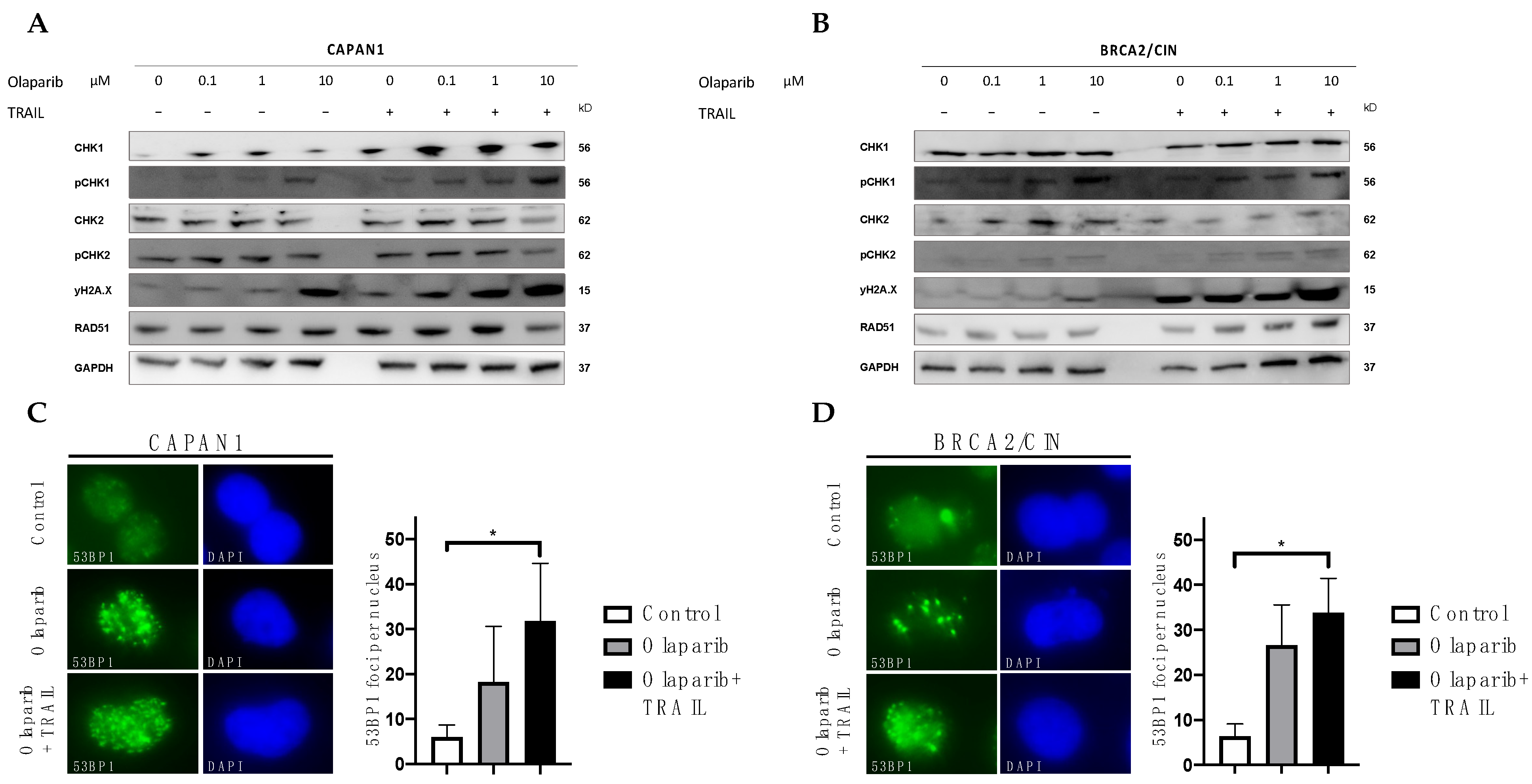
3.4. Influence of Olaparib and TRAIL on DNA Damage Repair
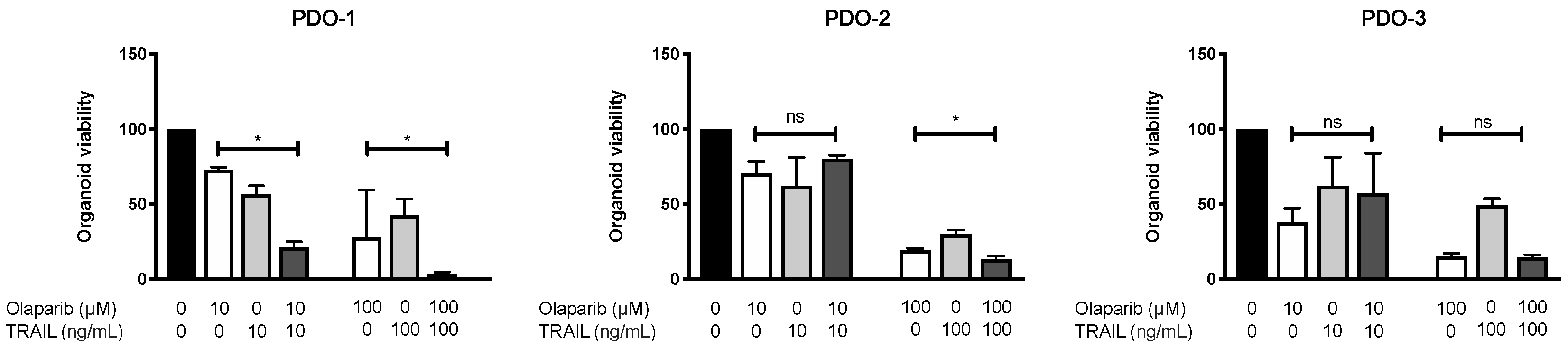
3.5. Combination of Olaparib and TRAIL Enhances Antitumor Activity in Patient-Derived Pancreatic Cancer Organoids
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomarkers Prev. 2016, 25, 16–27. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sohal, D.P.S.; Kennedy, E.B.; Cinar, P.; Conroy, T.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Lau, M.W.; Johnson, T.; Krishnamurthi, S.; et al. Metastatic Pancreatic Cancer: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 3217–3230. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.R.; Levine, D.A.; Tang, L.H.; Allen, P.J.; Jarnagin, W.; Brennan, M.F.; Offit, K.; Robson, M.E. BRCA Germline Mutations in Jewish Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2009, 27, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Thorlacius, S.; Olafsdottir, G.; Tryggvadottir, L.; Neuhausen, S.; Jonasson, J.G.; Tavtigian, S.V.; Tulinius, H.; Ögmundsdottir, H.M.; Eyfjörd, J.E. A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes. Nat. Genet. 1996, 13, 117–119. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Salo-Mullen, E.; Patil, S.M.; Pietanza, M.C.; Vijai, J.; Saloustros, E.; Hansen, N.A.; Kauff, N.D.; Kurtz, R.C.; Kelsen, D.P.; et al. Prevalence of BRCA1 and BRCA2 mutations in Ashkenazi Jewish families with breast and pancreatic cancer. Cancer 2012, 118, 493–499. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Patel, A.G.; Sarkaria, J.N.; Kaufmann, S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. USA 2011, 108, 3406–3411. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- European Medicines Agency. European Medicines Agency Medicines Database. Available online: https://www.ema.europa.eu/en/medicines (accessed on 20 April 2022).
- Food and Drug Administration Washington D.C., U.S. Drugs@fda. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm (accessed on 20 April 2022).
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Overall survival from the phase 3 POLO trial: Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. J. Clin. Oncol. 2021, 39, 378. [Google Scholar] [CrossRef]
- Karakashev, S.; Fukumoto, T.; Zhao, B.; Lin, J.; Wu, S.; Fatkhutdinov, N.; Park, P.H.; Semenova, G.; Jean, S.; Cadungog, M.G.; et al. EZH2 Inhibition Sensitizes CARM1-High, Homologous Recombination Proficient Ovarian Cancers to PARP Inhibition. Cancer Cell 2020, 37, 157–167.e156. [Google Scholar] [CrossRef]
- Jaaks, P.; Coker, E.A.; Vis, D.J.; Edwards, O.; Carpenter, E.F.; Leto, S.M.; Dwane, L.; Sassi, F.; Lightfoot, H.; Barthorpe, S.; et al. Effective drug combinations in breast, colon and pancreatic cancer cells. Nature 2022, 603, 166–173. [Google Scholar] [CrossRef]
- Choi, J.-R.; Shin, K.S.; Choi, C.Y.; Kang, S.J. PARP1 regulates the protein stability and proapoptotic function of HIPK2. Cell Death Dis. 2016, 7, e2438. [Google Scholar] [CrossRef]
- Rudolph, J.; Jung, K.; Luger, K. Inhibitors of PARP: Number crunching and structure gazing. Proc. Natl. Acad. Sci. USA 2022, 119, e2121979119. [Google Scholar] [CrossRef]
- Wang, S.C.; Shao, R.; Pao, A.Y.; Zhang, S.; Hung, M.C.; Su, L.K. Inhibition of cancer cell growth by BRCA2. Cancer Res. 2002, 62, 1311–1314. [Google Scholar]
- Hucl, T.; Rago, C.; Gallmeier, E.; Brody, J.R.; Gorospe, M.; Kern, S.E. A syngeneic variance library for functional annotation of human variation: Application to BRCA2. Cancer Res. 2008, 68, 5023–5030. [Google Scholar] [CrossRef]
- De Toni, E.N.; Ziesch, A.; Rizzani, A.; Török, H.-P.; Hocke, S.; Lü, S.; Wang, S.-C.; Hucl, T.; Göke, B.; Bruns, C.; et al. Inactivation of BRCA2 in human cancer cells identifies a subset of tumors with enhanced sensitivity towards death receptor-mediated apoptosis. Oncotarget 2016, 7, 9477–9490. [Google Scholar] [CrossRef]
- Mahajan, U.M.; Li, Q.; Alnatsha, A.; Maas, J.; Orth, M.; Maier, S.H.; Peterhansl, J.; Regel, I.; Sendler, M.; Wagh, P.R.; et al. Tumor-Specific Delivery of 5-Fluorouracil–Incorporated Epidermal Growth Factor Receptor–Targeted Aptamers as an Efficient Treatment in Pancreatic Ductal Adenocarcinoma Models. Gastroenterology 2021, 161, 996–1010.e1011. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Ye, L.; Mayerle, J.; Ziesch, A.; Reiter, F.P.; Gerbes, A.L.; De Toni, E.N. The PI3K inhibitor copanlisib synergizes with sorafenib to induce cell death in hepatocellular carcinoma. Cell Death Discov. 2019, 5, 86. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Deer, E.L.; González-Hernández, J.; Coursen, J.D.; Shea, J.E.; Ngatia, J.; Scaife, C.L.; Firpo, M.A.; Mulvihill, S.J. Phenotype and genotype of pancreatic cancer cell lines. Pancreas 2010, 39, 425–435. [Google Scholar] [CrossRef]
- Edwards, S.L.; Brough, R.; Lord, C.J.; Natrajan, R.; Vatcheva, R.; Levine, D.A.; Boyd, J.; Reis-Filho, J.S.; Ashworth, A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008, 451, 1111–1115. [Google Scholar] [CrossRef]
- Goggins, M.; Schutte, M.; Lu, J.; Moskaluk, C.A.; Weinstein, C.L.; Petersen, G.M.; Yeo, C.J.; Jackson, C.E.; Lynch, H.T.; Hruban, R.H.; et al. Germline BRCA2 gene mutations in patients with apparently sporadic pancreatic carcinomas. Cancer Res. 1996, 56, 5360–5364. [Google Scholar] [PubMed]
- Hinz, J.M. Role of homologous recombination in DNA interstrand crosslink repair. Environ. Mol. Mutagen. 2010, 51, 582–603. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Cui, T.Y.; Jasin, M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001, 61, 4842–4850. [Google Scholar]
- Yun, J.; Zhong, Q.; Kwak, J.-Y.; Lee, W.-H. Hypersensitivity of Brca1-deficient MEF to the DNA interstrand crosslinking agent mitomycin C is associated with defect in homologous recombination repair and aberrant S-phase arrest. Oncogene 2005, 24, 4009–4016. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Investig. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Soria, J.C.; Smit, E.; Khayat, D.; Besse, B.; Yang, X.; Hsu, C.P.; Reese, D.; Wiezorek, J.; Blackhall, F. Phase 1b study of dulanermin (recombinant human Apo2L/TRAIL) in combination with paclitaxel, carboplatin, and bevacizumab in patients with advanced non-squamous non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 1527–1533. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Messersmith, W.A.; Peddi, P.F.; Kapp, A.V.; Ashkenazi, A.; Royer-Joo, S.; Portera, C.C.; Kozloff, M.F. A phase 1B study of dulanermin in combination with modified FOLFOX6 plus bevacizumab in patients with metastatic colorectal cancer. Clin. Colorectal Cancer 2013, 12, 248–254. [Google Scholar] [CrossRef]
- Herbst, R.S.; Eckhardt, S.G.; Kurzrock, R.; Ebbinghaus, S.; O’Dwyer, P.J.; Gordon, M.S.; Novotny, W.; Goldwasser, M.A.; Tohnya, T.M.; Lum, B.L.; et al. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J. Clin. Oncol. 2010, 28, 2839–2846. [Google Scholar] [CrossRef]
- Soria, J.C.; Márk, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhász, E.; Pujol, J.L.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef]
- Ciuleanu, T.; Bazin, I.; Lungulescu, D.; Miron, L.; Bondarenko, I.; Deptala, A.; Rodriguez-Torres, M.; Giantonio, B.; Fox, N.L.; Wissel, P.; et al. A randomized, double-blind, placebo-controlled phase II study to assess the efficacy and safety of mapatumumab with sorafenib in patients with advanced hepatocellular carcinoma. Ann. Oncol. 2016, 27, 680–687. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Krzakowski, M.; Chmielowska, E.; Sebastian, M.; Hadler, D.; Fox, T.; Wang, Q.; Greenberg, J.; Beckman, R.A.; von Pawel, J. A randomized, double-blind, placebo-controlled phase 2 study of tigatuzumab (CS-1008) in combination with carboplatin/paclitaxel in patients with chemotherapy-naïve metastatic/unresectable non-small cell lung cancer. Lung Cancer 2013, 82, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Von Pawel, J.; Harvey, J.H.; Spigel, D.R.; Dediu, M.; Reck, M.; Cebotaru, C.L.; Humphreys, R.C.; Gribbin, M.J.; Fox, N.L.; Camidge, D.R. Phase II trial of mapatumumab, a fully human agonist monoclonal antibody to tumor necrosis factor-related apoptosis-inducing ligand receptor 1 (TRAIL-R1), in combination with paclitaxel and carboplatin in patients with advanced non-small-cell lung cancer. Clin. Lung Cancer 2014, 15, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Shi, M.; Jie, F.; Bai, Y.; Shen, P.; Yu, Z.; Wang, X.; Huang, C.; Tao, M.; Wang, Z.; et al. Phase III study of dulanermin (recombinant human tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand) combined with vinorelbine and cisplatin in patients with advanced non-small-cell lung cancer. Investig. New Drugs 2018, 36, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.W.; Koh, B.D.; Zhang, J.S.; Flatten, K.S.; Schneider, P.A.; Billadeau, D.D.; Hess, A.D.; Smith, B.D.; Karp, J.E.; Kaufmann, S.H. Poly(ADP-ribose) polymerase inhibitors sensitize cancer cells to death receptor-mediated apoptosis by enhancing death receptor expression. J. Biol. Chem. 2014, 289, 20543–20558. [Google Scholar] [CrossRef]
- Jungmichel, S.; Rosenthal, F.; Altmeyer, M.; Lukas, J.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 52, 272–285. [Google Scholar] [CrossRef]
- Carley, A.C.; Jalan, M.; Subramanyam, S.; Roy, R.; Borgstahl, G.E.O.; Powell, S.N. Replication Protein A Phosphorylation Facilitates RAD52-Dependent Homologous Recombination in BRCA-Deficient Cells. Mol. Cell. Biol. 2022, 42, 521–524. [Google Scholar] [CrossRef]
- Soniat, M.M.; Myler, L.R.; Kuo, H.C.; Paull, T.T.; Finkelstein, I.J. RPA Phosphorylation Inhibits DNA Resection. Mol Cell 2019, 75, 145–153.e145. [Google Scholar] [CrossRef]
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol. Life Sci. 2013, 70, 4009–4021. [Google Scholar] [CrossRef]
- Driehuis, E.; Hoeck, A.v.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Moore, K.N.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; et al. 811MO Maintenance olaparib for patients (pts) with newly diagnosed, advanced ovarian cancer (OC) and a BRCA mutation (BRCAm): 5-year (y) follow-up (f/u) from SOLO1. Ann. Oncol. 2020, 31, S613. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Oehm, A.; Behrmann, I.; Falk, W.; Pawlita, M.; Maier, G.; Klas, C.; Li-Weber, M.; Richards, S.; Dhein, J.; Trauth, B.C.; et al. Purification and molecular cloning of the APO-1 cell surface antigen, a member of the tumor necrosis factor/nerve growth factor receptor superfamily. Sequence identity with the Fas antigen. J. Biol. Chem. 1992, 267, 10709–10715. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.R.; van der Sloot, A.M.; Szegezdi, E.; Natoni, A.; Tur, V.; Cool, R.H.; Samali, A.; Serrano, L.; Quax, W.J. Enhancement of Antitumor Properties of rhTRAIL by Affinity Increase toward Its Death Receptors. Biochemistry 2009, 48, 2180–2191. [Google Scholar] [CrossRef]
- Reis, C.R.; van der Sloot, A.M.; Natoni, A.; Szegezdi, E.; Setroikromo, R.; Meijer, M.; Sjollema, K.; Stricher, F.; Cool, R.H.; Samali, A.; et al. Rapid and efficient cancer cell killing mediated by high-affinity death receptor homotrimerizing TRAIL variants. Cell Death Dis. 2010, 1, e83-e83. [Google Scholar] [CrossRef]
- Nair Pradeep, M.; Flores, H.; Gogineni, A.; Marsters, S.; Lawrence David, A.; Kelley Robert, F.; Ngu, H.; Sagolla, M.; Komuves, L.; Bourgon, R.; et al. Enhancing the antitumor efficacy of a cell-surface death ligand by covalent membrane display. Proc. Natl. Acad. Sci. USA 2015, 112, 5679–5684. [Google Scholar] [CrossRef]
- Wilson, N.S.; Yang, B.; Yang, A.; Loeser, S.; Marsters, S.; Lawrence, D.; Li, Y.; Pitti, R.; Totpal, K.; Yee, S.; et al. An Fcγ receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell 2011, 19, 101–113. [Google Scholar] [CrossRef]
- Papadopoulos, K.P.; Isaacs, R.; Bilic, S.; Kentsch, K.; Huet, H.A.; Hofmann, M.; Rasco, D.; Kundamal, N.; Tang, Z.; Cooksey, J.; et al. Unexpected hepatotoxicity in a phase I study of TAS266, a novel tetravalent agonistic Nanobody® targeting the DR5 receptor. Cancer Chemother. Pharmacol. 2015, 75, 887–895. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Gore, I.; Fong, L.; Venook, A.; Beck, S.B.; Dorazio, P.; Criscitiello, P.J.; Healey, D.I.; Huang, B.; Gomez-Navarro, J.; et al. Phase II study of the anti-cytotoxic T-lymphocyte-associated antigen 4 monoclonal antibody, tremelimumab, in patients with refractory metastatic colorectal cancer. J. Clin. Oncol. 2010, 28, 3485–3490. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: Safety, clinical activity, pharmacodynamics, and immunologic correlates. J. Clin. Oncol. 2010, 28, 3167–3175. [Google Scholar] [CrossRef]
- Kojima, Y.; Nakayama, M.; Nishina, T.; Nakano, H.; Koyanagi, M.; Takeda, K.; Okumura, K.; Yagita, H. Importin β1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J. Biol. Chem. 2011, 286, 43383–43393. [Google Scholar] [CrossRef] [PubMed]
- Gallmeier, E.; Bader, D.C.; Kriegl, L.; Berezowska, S.; Seeliger, H.; Göke, B.; Kirchner, T.; Bruns, C.; De Toni, E.N. Loss of TRAIL-receptors is a recurrent feature in pancreatic cancer and determines the prognosis of patients with no nodal metastasis after surgery. PLoS ONE 2013, 8, e56760. [Google Scholar] [CrossRef] [PubMed]
- Lister, A.; Nedjadi, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef]
- Arlt, A.; Sebens, S.; Krebs, S.; Geismann, C.; Grossmann, M.; Kruse, M.L.; Schreiber, S.; Schäfer, H. Inhibition of the Nrf2 transcription factor by the alkaloid trigonelline renders pancreatic cancer cells more susceptible to apoptosis through decreased proteasomal gene expression and proteasome activity. Oncogene 2013, 32, 4825–4835. [Google Scholar] [CrossRef]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: Implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Singh, A.; Boldin-Adamsky, S.; Thimmulappa, R.K.; Rath, S.K.; Ashush, H.; Coulter, J.; Blackford, A.; Goodman, S.N.; Bunz, F.; Watson, W.H.; et al. RNAi-mediated silencing of nuclear factor erythroid-2-related factor 2 gene expression in non-small cell lung cancer inhibits tumor growth and increases efficacy of chemotherapy. Cancer Res. 2008, 68, 7975–7984. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, J.; Liu, S.; Huang, Y.; Chen, B.; Wang, D. Nrf2 knockdown by shRNA inhibits tumor growth and increases efficacy of chemotherapy in cervical cancer. Cancer Chemother. Pharmacol. 2012, 69, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Schumacher, S.; Rosskopf, R.; Rhein, T.; Schmidt-Petersen, F.; Gatzemeier, F.; Haag, F.; Borgmann, K.; Willers, H.; Dahm-Daphi, J. Hierarchy of nonhomologous end-joining, single-strand annealing and gene conversion at site-directed DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 4088–4098. [Google Scholar] [CrossRef] [PubMed]
- Moyal, L.; Lerenthal, Y.; Gana-Weisz, M.; Mass, G.; So, S.; Wang, S.Y.; Eppink, B.; Chung, Y.M.; Shalev, G.; Shema, E.; et al. Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 2011, 41, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef]
- Schulte-Uentrop, L.; El-Awady, R.A.; Schliecker, L.; Willers, H.; Dahm-Daphi, J. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 2561–2569. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ben Khaled, N.; Hammer, K.; Ye, L.; Alnatsha, A.; Widholz, S.A.; Piseddu, I.; Sirtl, S.; Schneider, J.; Munker, S.; Mahajan, U.M.; et al. TRAIL Receptor Targeting Agents Potentiate PARP Inhibitor Efficacy in Pancreatic Cancer Independently of BRCA2 Mutation Status. Cancers 2022, 14, 5240. https://doi.org/10.3390/cancers14215240
Ben Khaled N, Hammer K, Ye L, Alnatsha A, Widholz SA, Piseddu I, Sirtl S, Schneider J, Munker S, Mahajan UM, et al. TRAIL Receptor Targeting Agents Potentiate PARP Inhibitor Efficacy in Pancreatic Cancer Independently of BRCA2 Mutation Status. Cancers. 2022; 14(21):5240. https://doi.org/10.3390/cancers14215240
Chicago/Turabian StyleBen Khaled, Najib, Katharina Hammer, Liangtao Ye, Ahmed Alnatsha, Sebastian A. Widholz, Ignazio Piseddu, Simon Sirtl, Julia Schneider, Stefan Munker, Ujjwal Mukund Mahajan, and et al. 2022. "TRAIL Receptor Targeting Agents Potentiate PARP Inhibitor Efficacy in Pancreatic Cancer Independently of BRCA2 Mutation Status" Cancers 14, no. 21: 5240. https://doi.org/10.3390/cancers14215240
APA StyleBen Khaled, N., Hammer, K., Ye, L., Alnatsha, A., Widholz, S. A., Piseddu, I., Sirtl, S., Schneider, J., Munker, S., Mahajan, U. M., Montero, J. J., Griger, J., Mayerle, J., Reiter, F. P., & De Toni, E. N. (2022). TRAIL Receptor Targeting Agents Potentiate PARP Inhibitor Efficacy in Pancreatic Cancer Independently of BRCA2 Mutation Status. Cancers, 14(21), 5240. https://doi.org/10.3390/cancers14215240