






Cells 2026, 15(11), 1005; https://doi.org/10.3390/cells15111005 (registering DOI) - 29 May 2026
Abstract
Rheumatoid arthritis (RA) is a systemic immune-related disease characterized by chronic synovial inflammation and progressive joint destruction. However, the molecular mechanisms and diagnostic biomarkers underlying RA remain unclear. In this study, we aimed to identify potential biomarkers for clinical diagnosis of RA and
[...] Read more.
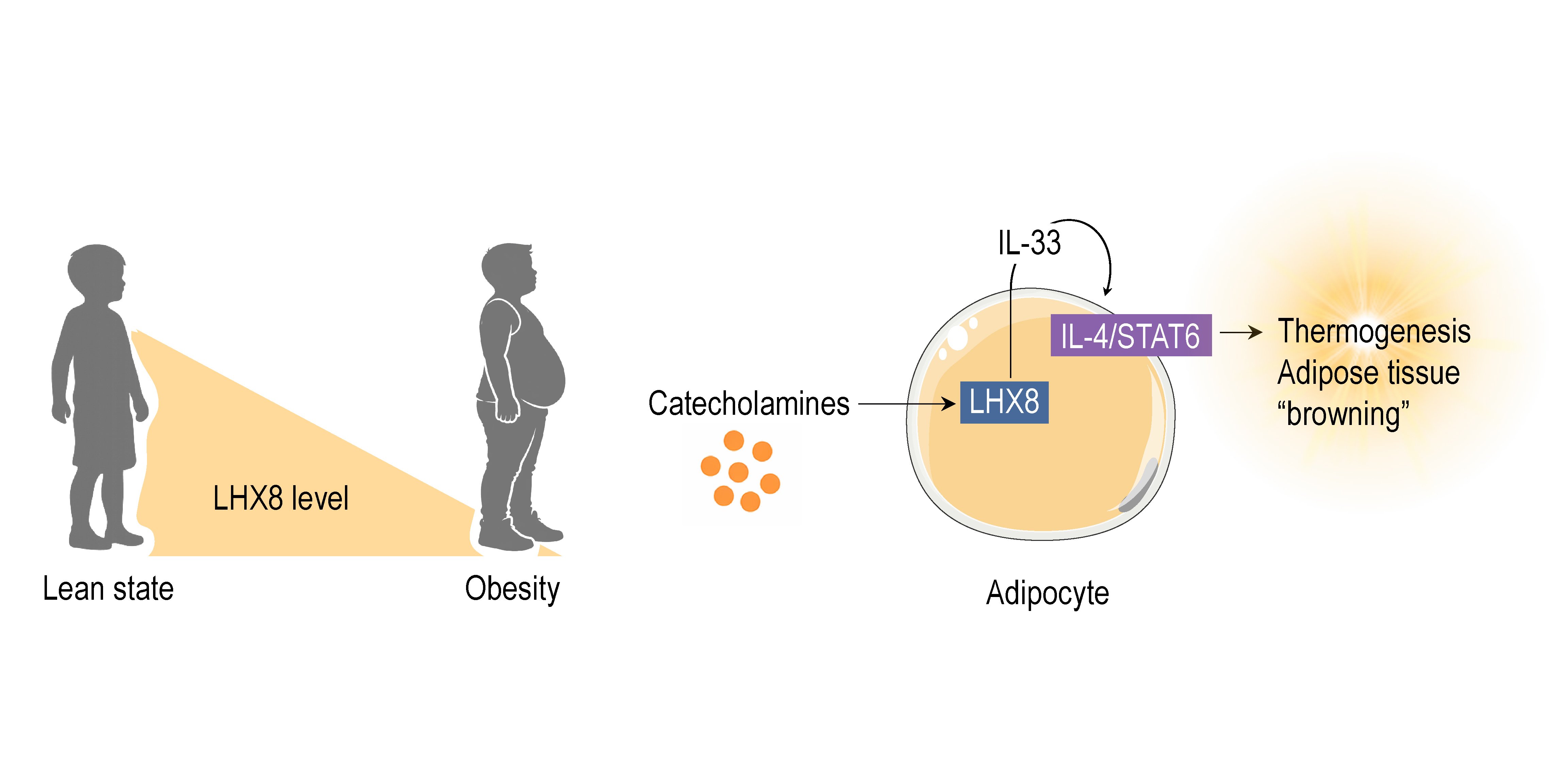
Rheumatoid arthritis (RA) is a systemic immune-related disease characterized by chronic synovial inflammation and progressive joint destruction. However, the molecular mechanisms and diagnostic biomarkers underlying RA remain unclear. In this study, we aimed to identify potential biomarkers for clinical diagnosis of RA and to investigate their association with immune infiltration. By integrating differentially expressed genes analysis (DEGs) and weighted gene co-expression network analysis (WGCNA), we identified PSMB9 as a hub gene associated with RA. Gene set enrichment analysis (GSEA) and immune infiltration analysis revealed a strong association between RA and macrophage infiltration. Single-cell RNA sequencing datasets also suggest that PSMB9 is not only highly expressed in macrophage but is also present in synovial cells. We employed cellular thermal shift assay (CETSA) combined with Western blot to validate the interaction between sinomenine (SIN) and the target protein. CETSA results demonstrated that, compared with the control group, SIN increased the thermal stability of PSMB9, suggesting direct binding between the two. Western blot experiments further confirmed that PSMB9 protein expression was significantly downregulated following SIN treatment. PSMB9 may serve as potential diagnostic biomarker and therapeutic targets for RA. Moreover, our data suggest SIN may exert anti-inflammatory effects through regulation of PSMB9. This study also provides an additional insight into the underlying mechanisms involved in the progression of RA and discover a new molecular target for SIN.
Full article
(This article belongs to the Special Issue Study on Immune Activity of Natural Products)
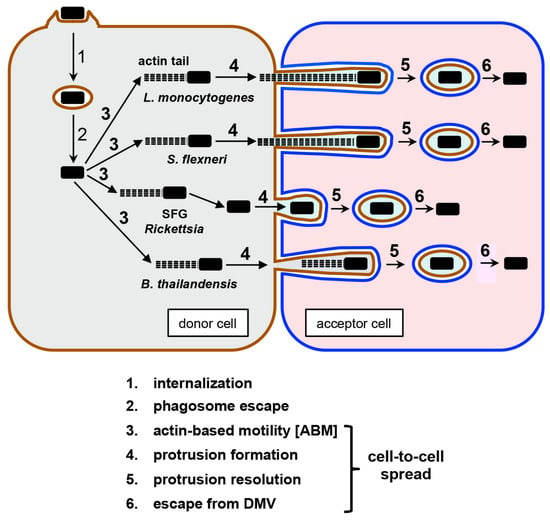
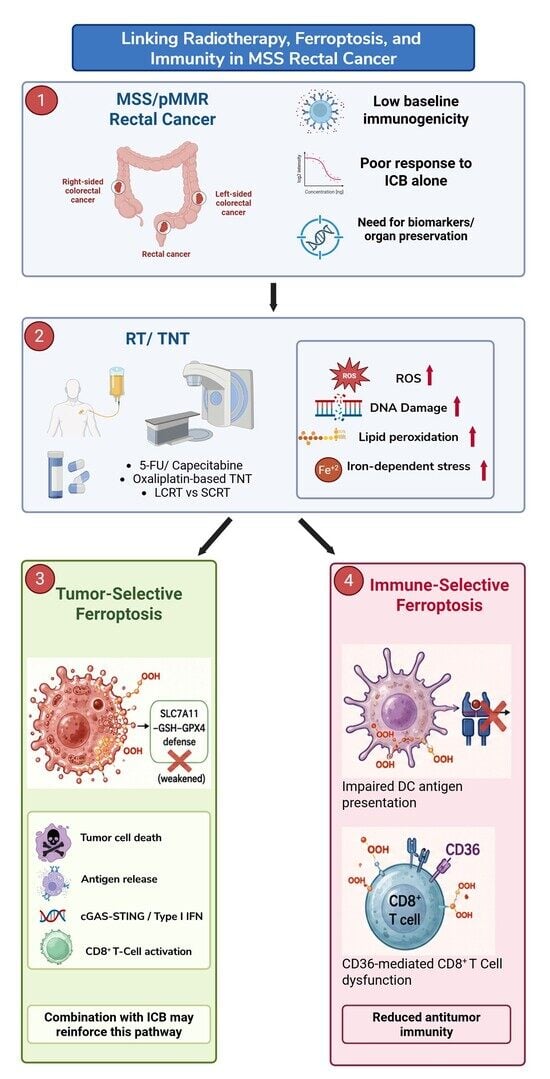
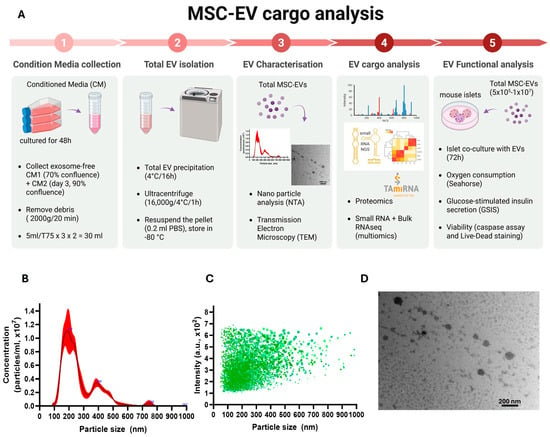

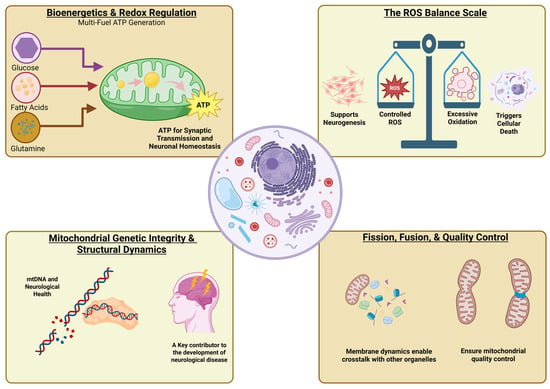
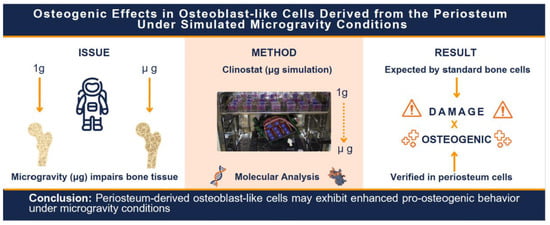
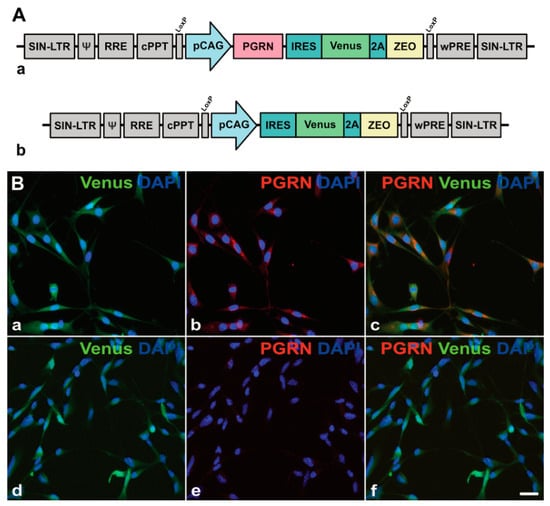
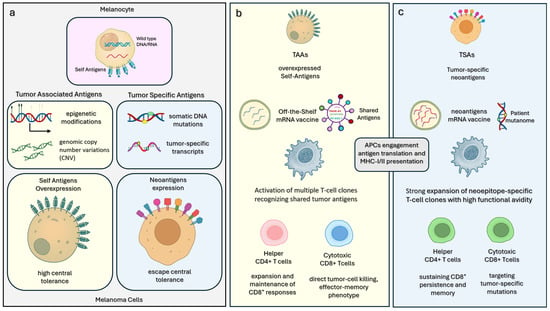





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}