
Anti-Anaplastic Thyroid Cancer (ATC) Effects and Mechanisms of PLX3397 (Pexidartinib), a Multi-Targeted Tyrosine Kinase Inhibitor (TKI)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Culturing, and Chemicals
2.2. CCK-8 for Cell Viability Determination
2.3. LDH Cell Cytotoxicity Assay
2.4. Plate Cloning Assay
2.5. EdU (5-Ethynyl-2′-deoxyuridine) Assay
2.6. Apoptotic Cell Detection
2.7. Evaluating Caspase-3 Activity
2.8. Cell Cycle Evaluation
2.9. ROS Determination
2.10. Immunofluorescence (IF)
2.11. Cell Transfection
2.12. Western Blot Assay
2.13. Xenografts and Immunohistochemistry
2.14. Statistical Analysis
3. Results
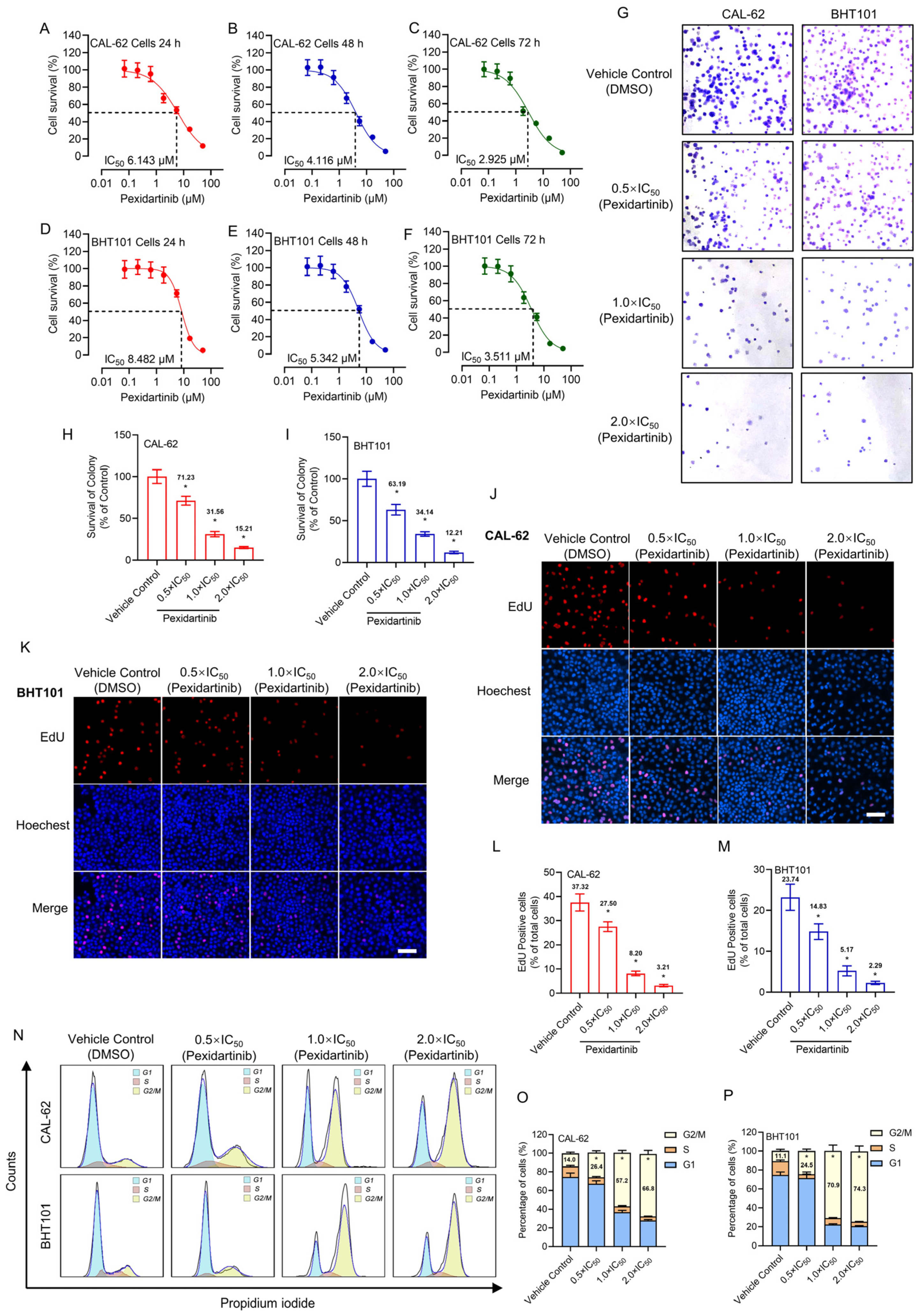
3.1. Pexidartinib Antagonizes ATC Cell Proliferation
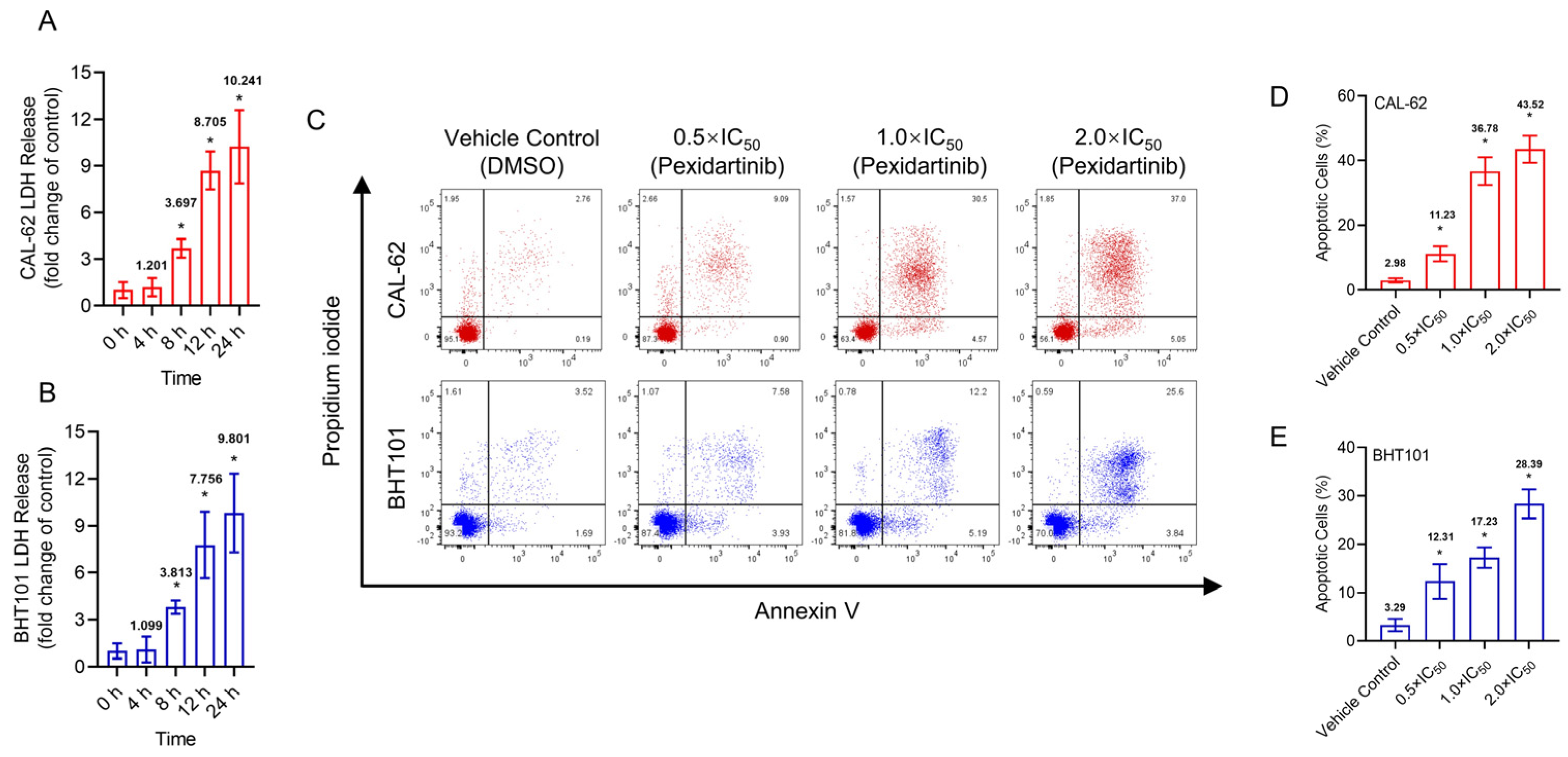
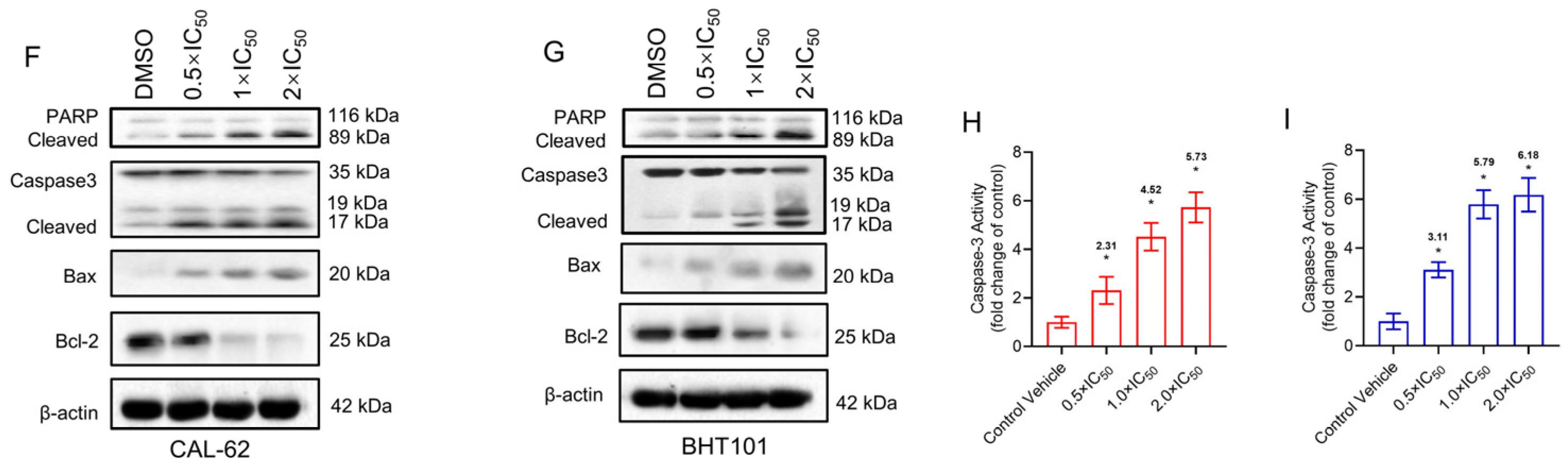
3.2. Pexidartinib Illustrates Cytotoxicity Effects and Increases Apoptotic Cells in ATC
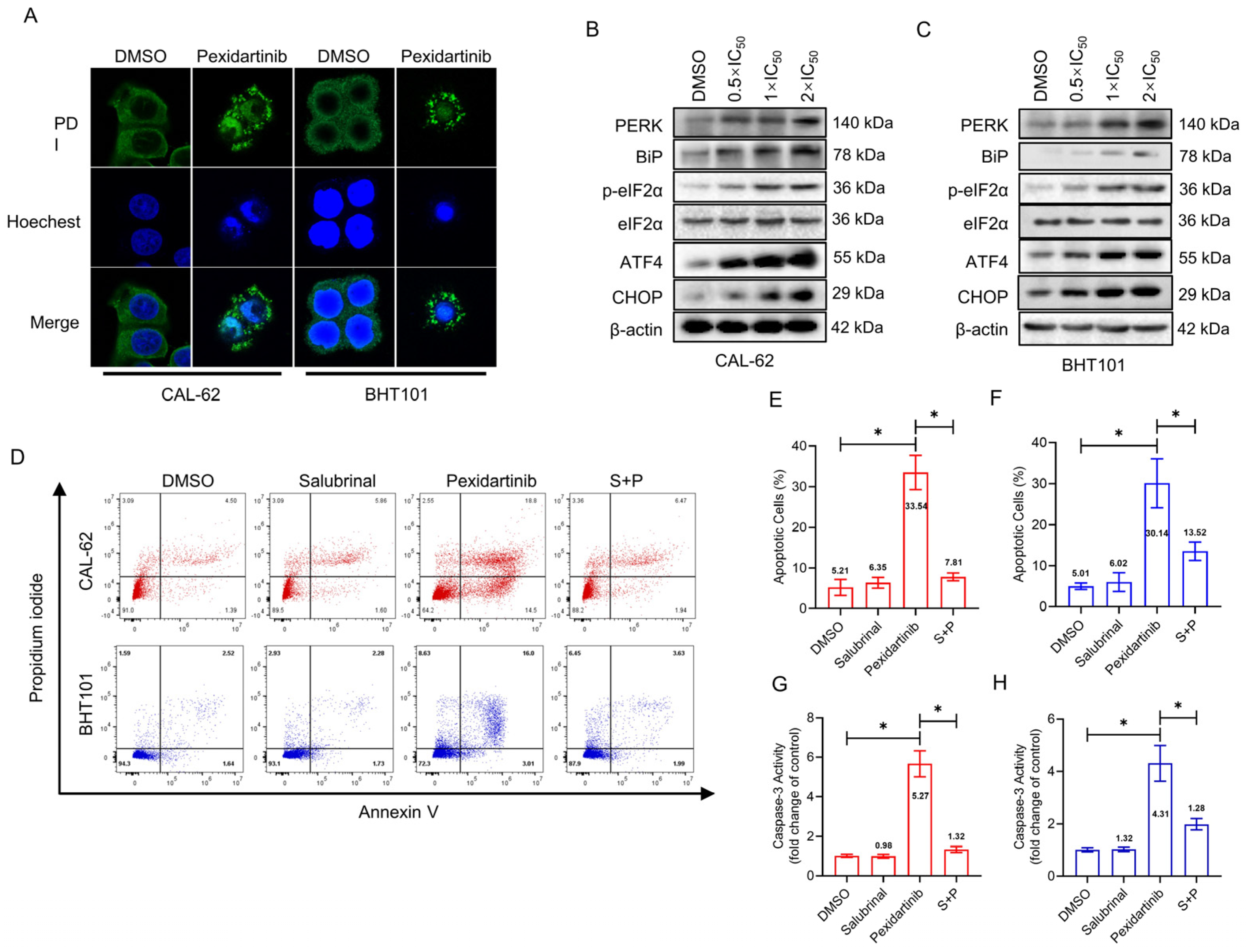
3.3. ER Stress Is Responsible for the Apoptosis Induced by Pexidartinib
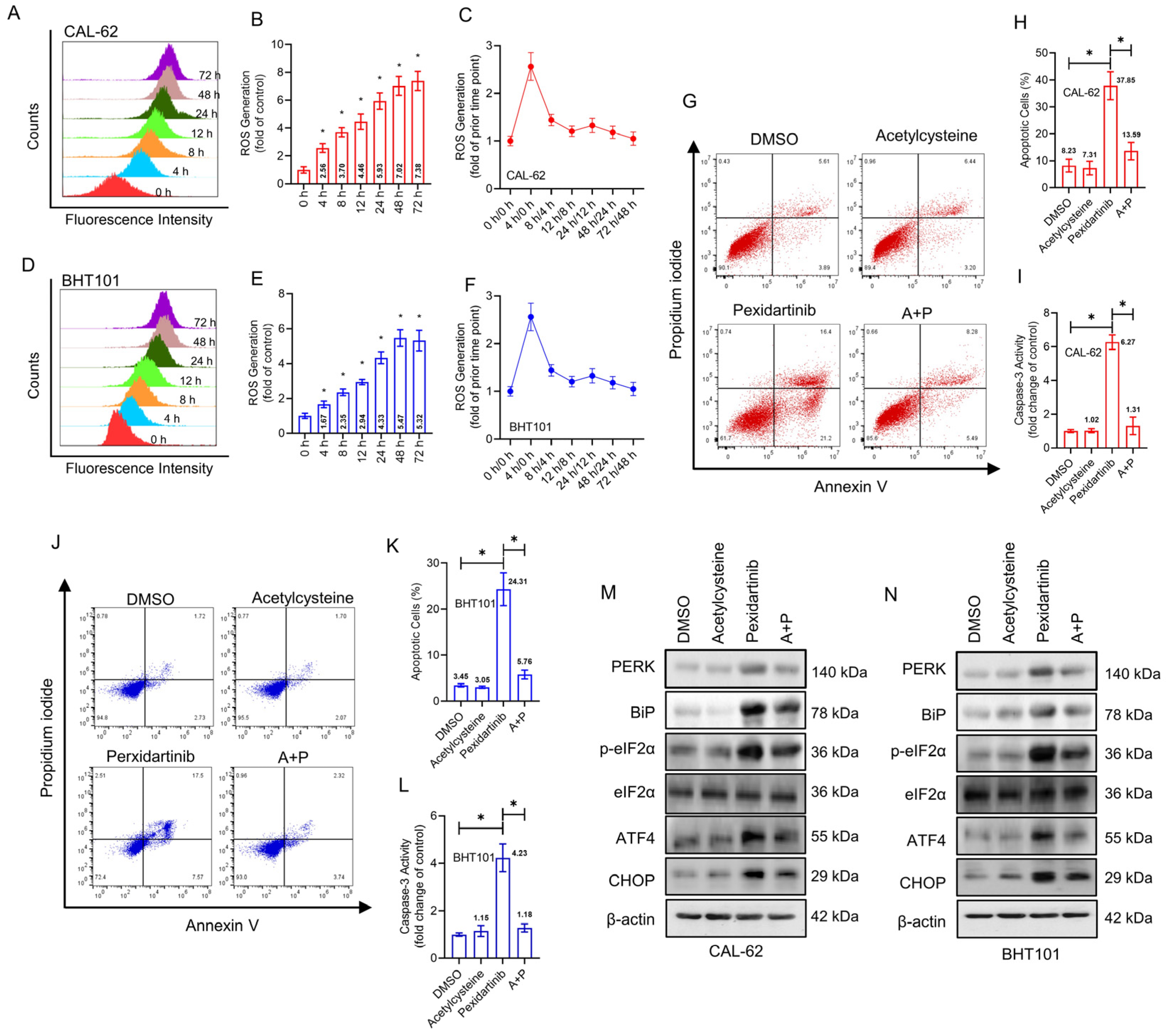
3.4. Pexidartinib Induces ER Stress and Accompanying Apoptosis in ATC Cells by Elevation of Intracellular ROS
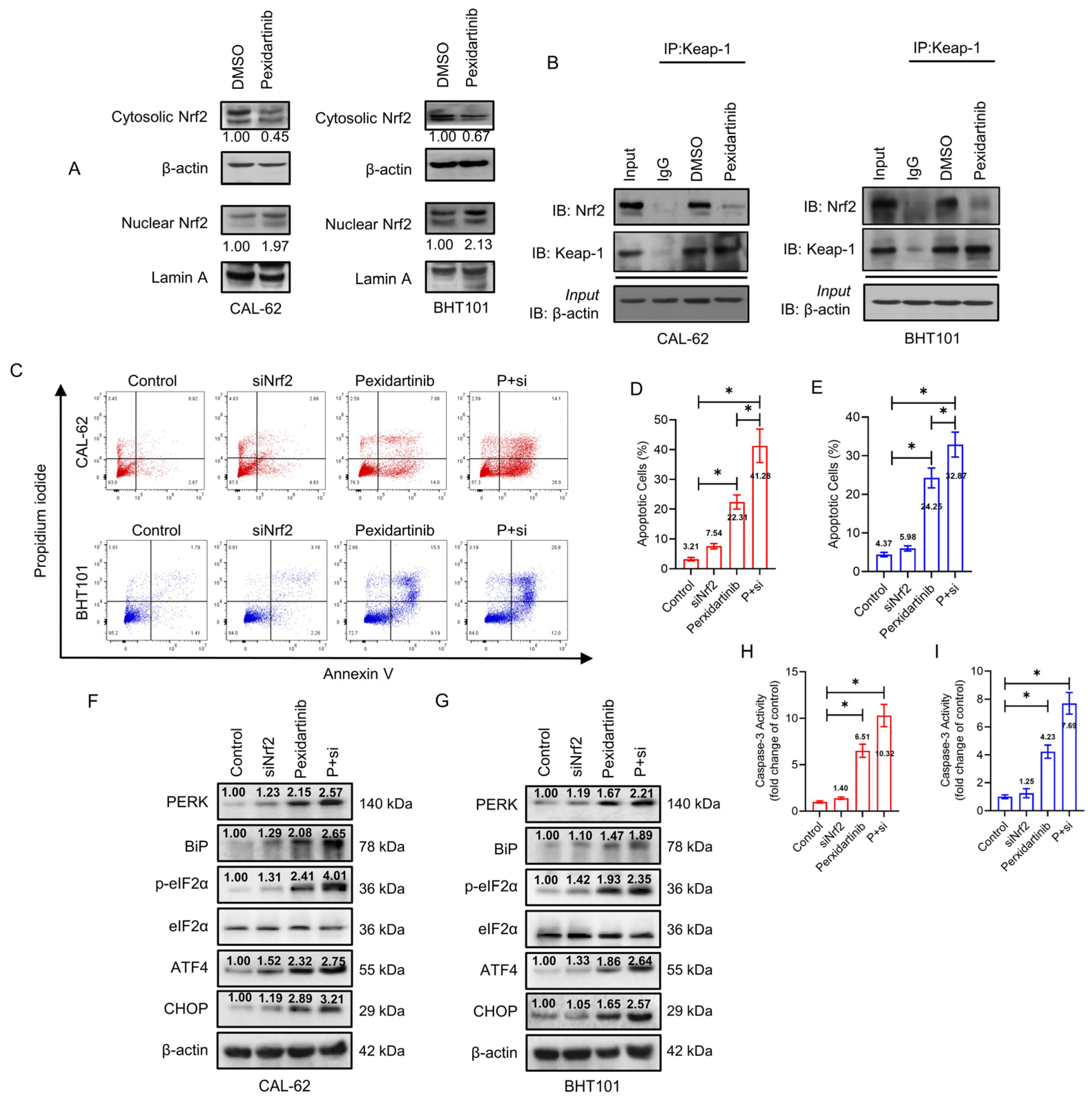
3.5. Knockdown of Nrf2 Elevates Anticancer Activity of Pexidartinib in ATC Cells
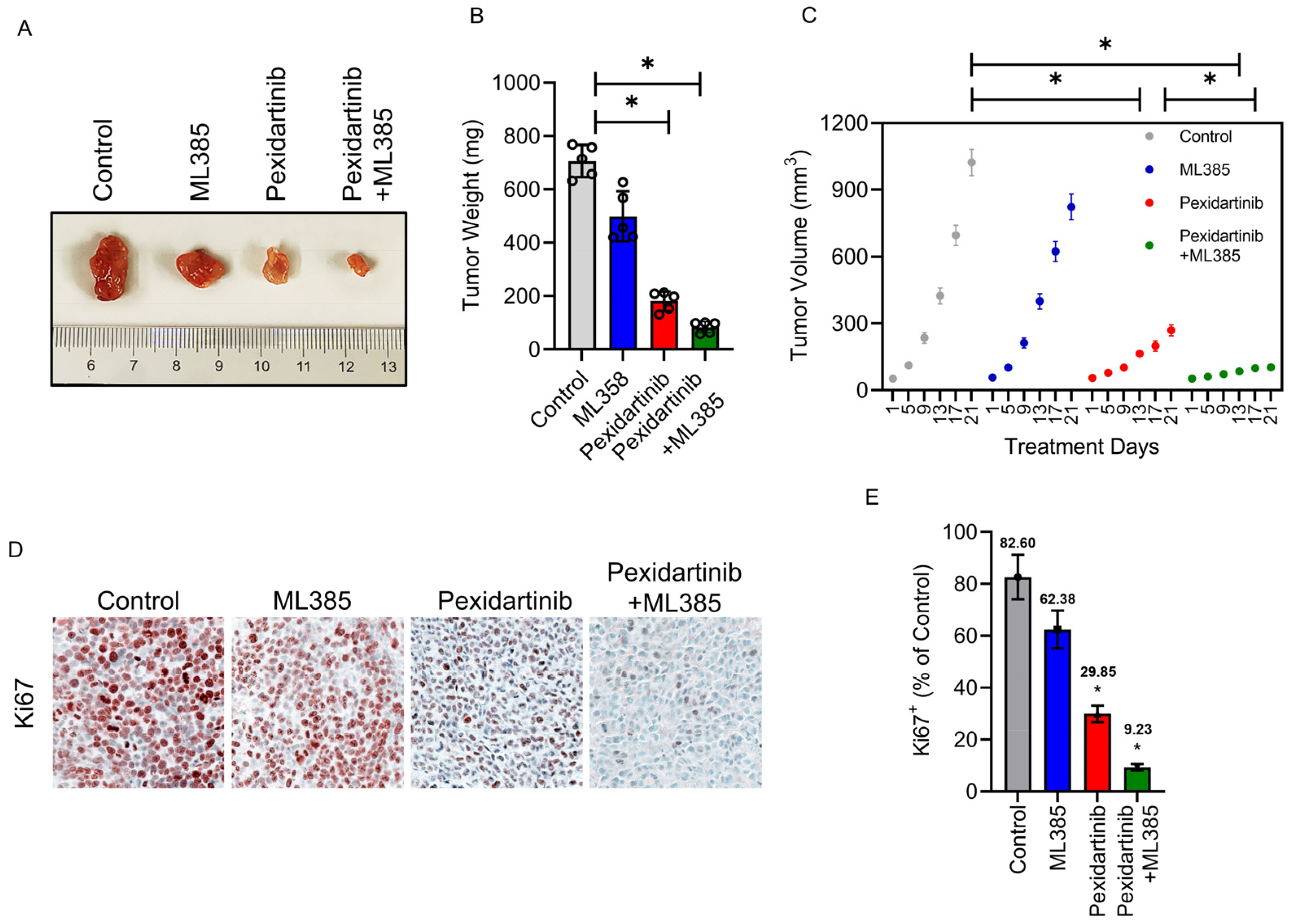
3.6. Pexidartinib Inhibits ATC Tumor Growth In Vivo, Co-Administration with Nrf2 Inhibitor Enhance the Anti-ATC Effects of Pexidartinib in Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bible, K.C.; Kebebew, E.; Brierley, J.; Brito, J.P.; Cabanillas, M.E.; Clark, T.J., Jr.; Di Cristofano, A.; Foote, R.; Giordano, T.; Kasperbauer, J.; et al. 2021 American Thyroid Association Guidelines for Management of Patients with Anaplastic Thyroid Cancer. Thyroid 2021, 31, 337–386. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Tulla, K.; Maker, A.V.; Burman, K.D.; Prabhakar, B.S. Therapeutic advances in anaplastic thyroid cancer: A current perspective. Mol. Cancer 2018, 17, 154. [Google Scholar] [CrossRef] [PubMed]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef]
- Feng, H.; Cheng, X.; Kuang, J.; Chen, L.; Yuen, S.; Shi, M.; Liang, J.; Shen, B.; Jin, Z.; Yan, J.; et al. Apatinib-induced protective autophagy and apoptosis through the AKT-mTOR pathway in anaplastic thyroid cancer. Cell Death Dis. 2018, 9, 1030. [Google Scholar] [CrossRef] [PubMed]
- Ao, J.-Y.; Zhu, X.-D.; Chai, Z.-T.; Cai, H.; Zhang, Y.-Y.; Zhang, K.-Z.; Kong, L.-Q.; Zhang, N.; Ye, B.-G.; Ma, D.-N. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular CarcinomaCSF1R Blockade and Macrophage Polarization. Mol. Cancer Ther. 2017, 16, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef]
- Tap, W.D.; Wainberg, Z.A.; Anthony, S.P.; Ibrahim, P.N.; Zhang, C.; Healey, J.H.; Chmielowski, B.; Staddon, A.P.; Cohn, A.L.; Shapiro, G.I. Structure-guided blockade of CSF1R kinase in tenosynovial giant-cell tumor. N. Engl. J. Med. 2015, 373, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef]
- Rosenbaum, E.; Kelly, C.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Movva, S.; Condy, M.; Adamson, T.; Mcfadyen, C.R. A phase I study of binimetinib (MEK162) combined with pexidartinib (PLX3397) in patients with advanced gastrointestinal stromal tumor. Oncologist 2019, 24, 1309-e983. [Google Scholar] [CrossRef]
- Wagner, A.J.; Tap, W.D.; Shields, A.F.; Patnaik, A.; Chugh, R.; Tinoco, G.; Michelson, G.; Alcantar, O.; Pelayo, M.; Zhang, C. A Phase I Pharmacokinetic (PK) and Pharmacodynamic (PD) Study of PLX9486 alone and in Combination (Combo) with the KIT Inhibitors Pexidartinib (Pexi) or Sunitinib (Su) in Patients (Pts) with Advanced Solid Tumors and Gastrointestinal Stromal Tumor (GIST). J. Clin. Oncol. 2018, 36, 11509. [Google Scholar] [CrossRef]
- Cassier, P.A.; Garin, G.; Eberst, L.; Delord, J.-P.; Chabaud, S.; Terret, C.; Montane, L.; Bidaux, A.-S.; Laurent, S.; Jaubert, L. MEDIPLEX: A Phase 1 Study of Durvalumab (D) Combined with Pexidartinib (P) in Patients (Pts) with Advanced Pancreatic Ductal Adenocarcinoma (PDAC) and Colorectal Cancer (CRC). J. Clin. Oncol. 2019, 37, 2579. [Google Scholar] [CrossRef]
- Hittson, L.; Glod, J.; Amaya, M.; Derdak, J.; Widemann, B.C.; Kaplan, R.N. Phase I Study of Pexidartinib (PLX3397) in Children with Refractory Leukemias and Solid Tumors Including Neurofibromatosis Type I (NF1) Related Plexiform Neurofibromas (PN). J. Clin. Oncol. 2017, 35, 10546. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Völz, R.; Harris, W.; Hirt, H.; Lee, Y.H. ROS homeostasis mediated by MPK4 and SUMM2 determines synergid cell death. Nat. Commun. 2022, 13, 1746. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387, 95–105. [Google Scholar] [CrossRef]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ou-Yang, F.; Hou, M.F.; Huang, H.W.; Wang, H.R.; Li, K.T.; Fayyaz, S.; Shu, C.W.; Chang, H.W. Oxidative stress-modulating drugs have preferential anticancer effects—Involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol. 2019, 58, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, X.; Sun, J.; Lei, Q.; Wang, Q.; Pan, D.; Ding, M.; Ding, Y. Reactive oxygen species mediate anlotinib-induced apoptosis via activation of endoplasmic reticulum stress in pancreatic cancer. Cell Death Dis. 2020, 11, 766. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.L.; Li, B.; Zhao, H.P.; Zhao, H.C.; Feng, X.Q. A function of fascin1 in the colony formation of mouse embryonic stem cells. Stem. Cells 2020, 38, 1078–1090. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.; Lin, M.; Zhang, F.; Hu, Y.; Xu, X.; Li, Y.; Liu, K.; Ma, X.; Tian, X.; Yao, J. Dietary flavonoid genistein induces Nrf2 and phase II detoxification gene expression via ERKs and PKC pathways and protects against oxidative stress in Caco-2 cells. Mol. Nutr. Food Res. 2013, 57, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liang, M.; Jiang, J.; He, R.; Wang, M.; Guo, X.; Shen, M.; Qin, R. Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int. J. Biol. Sci. 2018, 14, 1291–1305. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagari, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajghate, S.; et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Zhu, J.; Lee, W.K.; Altan-Bonnet, G.; Meltzer, P.; Cheng, S.Y. Inflammation suppression prevents tumor cell proliferation in a mouse model of thyroid cancer. Am. J. Cancer Res. 2020, 10, 1857–1870. [Google Scholar]
- Varghese, F.; Bukhari, A.B.; Malhotra, R.; De, A. IHC Profiler: An open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS ONE 2014, 9, e96801. [Google Scholar] [CrossRef]
- Kitahara, C.M.; Sosa, J.A. The changing incidence of thyroid cancer. Nat. Rev. Endocrinol. 2016, 12, 646–653. [Google Scholar] [CrossRef]
- Liao, Y.; Hua, Y.; Li, Y.; Zhang, C.; Yu, W.; Guo, P.; Zou, K.; Li, W.; Sun, Y.; Wang, R.; et al. CRSP8 promotes thyroid cancer progression by antagonizing IKKα-induced cell differentiation. Cell Death Differ. 2021, 28, 1347–1363. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Xian, M.; Cao, H.; Cao, J.; Shao, X.; Zhu, D.; Zhang, N.; Huang, P.; Li, W.; Yang, B.; Ying, M.; et al. Bortezomib sensitizes human osteosarcoma cells to adriamycin-induced apoptosis through ROS-dependent activation of p-eIF2α/ATF4/CHOP axis. Int. J. Cancer 2017, 141, 1029–1041. [Google Scholar] [CrossRef]
- Xu, J.; Ji, L.; Ruan, Y.; Wan, Z.; Lin, Z.; Xia, S.; Tao, L.; Zheng, J.; Cai, L.; Wang, Y.; et al. UBQLN1 mediates sorafenib resistance through regulating mitochondrial biogenesis and ROS homeostasis by targeting PGC1β in hepatocellular carcinoma. Signal. Transduct. Target Ther. 2021, 6, 190. [Google Scholar] [CrossRef]
- Li, Y.; Chen, C.; Liu, H.L.; Li, C.G.; Zhang, Z.F.; Wang, C.L. Pazopanib restricts small cell lung cancer proliferation via reactive oxygen species-mediated endoplasmic reticulum stress. Thorac. Cancer 2022, 13, 2421–2428. [Google Scholar] [CrossRef]
- Yu, J.; Li, T.; Liu, Y.; Wang, X.; Zhang, J.; Wang, X.; Shi, G.; Lou, J.; Wang, L.; Wang, C.C.; et al. Phosphorylation switches protein disulfide isomerase activity to maintain proteostasis and attenuate ER stress. EMBO J. 2020, 39, e103841. [Google Scholar] [CrossRef]
- Zhang, Y.; Swanda, R.V.; Nie, L.; Liu, X.; Wang, C.; Lee, H.; Lei, G.; Mao, C.; Koppula, P.; Cheng, W.; et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021, 12, 1589. [Google Scholar] [CrossRef]
- Colin, I.M.; Denef, J.F.; Lengelé, B.; Many, M.C.; Gérard, A.C. Recent insights into the cell biology of thyroid angiofollicular units. Endocr. Rev. 2013, 34, 209–238. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Wiel, C.; Le Gal, K.; Ibrahim, M.X.; Jahangir, C.A.; Kashif, M.; Yao, H.; Ziegler, D.V.; Xu, X.; Ghosh, T.; Mondal, T.; et al. BACH1 Stabilization by Antioxidants Stimulates Lung Cancer Metastasis. Cell 2019, 178, 330–345.e322. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.; Wang, H.; Xu, S.W.; Yang, L.H.; Chen, W.; Zhao, S.X.; Shen, H.; Liu, Q.; Yang, R.M.; Wang, J. Variants in oxidative stress-related genes affect the chemosensitivity through Nrf2-mediated signaling pathway in biliary tract cancer. EBioMedicine 2019, 48, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, J.; Wang, Y.; Zhao, L.; Wang, C.; Zhang, Z. Anti-Anaplastic Thyroid Cancer (ATC) Effects and Mechanisms of PLX3397 (Pexidartinib), a Multi-Targeted Tyrosine Kinase Inhibitor (TKI). Cancers 2023, 15, 172. https://doi.org/10.3390/cancers15010172
Luo J, Wang Y, Zhao L, Wang C, Zhang Z. Anti-Anaplastic Thyroid Cancer (ATC) Effects and Mechanisms of PLX3397 (Pexidartinib), a Multi-Targeted Tyrosine Kinase Inhibitor (TKI). Cancers. 2023; 15(1):172. https://doi.org/10.3390/cancers15010172
Chicago/Turabian StyleLuo, Jingtao, Yun Wang, Lingkun Zhao, Chunli Wang, and Ze Zhang. 2023. "Anti-Anaplastic Thyroid Cancer (ATC) Effects and Mechanisms of PLX3397 (Pexidartinib), a Multi-Targeted Tyrosine Kinase Inhibitor (TKI)" Cancers 15, no. 1: 172. https://doi.org/10.3390/cancers15010172
APA StyleLuo, J., Wang, Y., Zhao, L., Wang, C., & Zhang, Z. (2023). Anti-Anaplastic Thyroid Cancer (ATC) Effects and Mechanisms of PLX3397 (Pexidartinib), a Multi-Targeted Tyrosine Kinase Inhibitor (TKI). Cancers, 15(1), 172. https://doi.org/10.3390/cancers15010172