


Viruses, Volume 11, Issue 2 (February 2019) – 107 articles
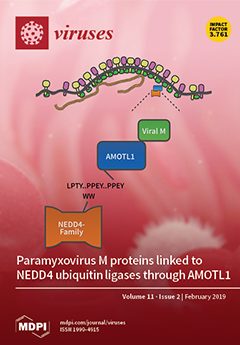
Cover Story (view full-size image):
Many enveloped viruses employ late domains such as PPXY to recruit the host ESCRT machinery needed for budding and particle release. Paramyxoviruses typically lack late domain sequences, yet budding of these viruses is often ESCRT-dependent. Here, we provide evidence for a model in which paramyxoviruses use AMOTL1 as a linker to indirectly recruit the same WW domain-containing NEDD4 ubiquitin ligases for budding that other enveloped viruses recruit directly through PPXY late domains. View this paper.
- Issues are regarded as officially published after their release is announced to the table of contents alert mailing list.
- You may sign up for e-mail alerts to receive table of contents of newly released issues.
- PDF is the official format for papers published in both, html and pdf forms. To view the papers in pdf format, click on the "PDF Full-text" link, and use the free Adobe Reader to open them.
Previous Issue
Next Issue