







Viruses 2026, 18(8), 846; https://doi.org/10.3390/v18080846 (registering DOI) - 1 Aug 2026
Abstract
Human norovirus (HuNoV) is one of the most important foodborne viruses and poses a significant risk in frozen foods along the cold chain. Traditional disinfectants exhibit reduced efficacy in frozen matrices and often fail to meet the requirements for food applications. This study
[...] Read more.
Human norovirus (HuNoV) is one of the most important foodborne viruses and poses a significant risk in frozen foods along the cold chain. Traditional disinfectants exhibit reduced efficacy in frozen matrices and often fail to meet the requirements for food applications. This study optimized and evaluated a novel antifreeze–peracetic acid formulation (Af-PAA) for application on frozen foods, achieving disinfection performance at −40 °C. The formulation was optimized using four indicator microorganisms, with effective Af-PAA concentrations of 200 mg/L for Escherichia coli, 200 mg/L for Staphylococcus aureus, 500 mg/L for Candida albicans, and 2000 mg/L for Aspergillus niger. HuNoV GII.4[P31] propagated in zebrafish was inoculated onto frozen food samples. The frozen blueberries (500 mg/L), carrots (500 mg/L), chicken breast (2000 mg/L), and Arctic shrimp (2000 mg/L) were then subjected to Af-PAA treatment at −40 °C for 60 min with 60 s vortex mixing. RNase-RT-qPCR analysis showed a 3.0–3.1 log10 reduction in RNase-resistant HuNoV RNA signals across all tested food under the corresponding treatment concentrations. No visual changes were observed in treated foods. This study demonstrated that Af-PAA met tested food disinfectant requirements and effectively inactivated indicator microorganisms. HuNoV signals were also markedly reduced on the tested foods. These findings support its application for microbial risk control in cold-chain foods.
Full article
(This article belongs to the Special Issue Enteric Viruses in Environment and Humans: Identification, Surveillance and Control)
►
Show Figures
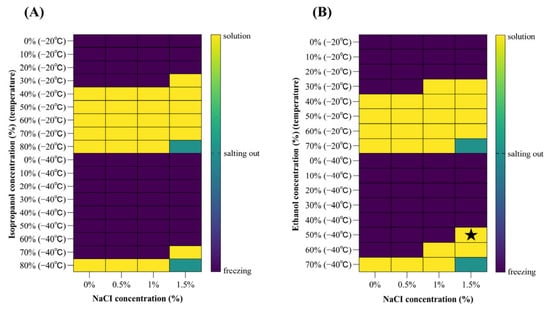
Figure 1










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}