Novel Approaches for The Development of Live Attenuated Influenza Vaccines



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
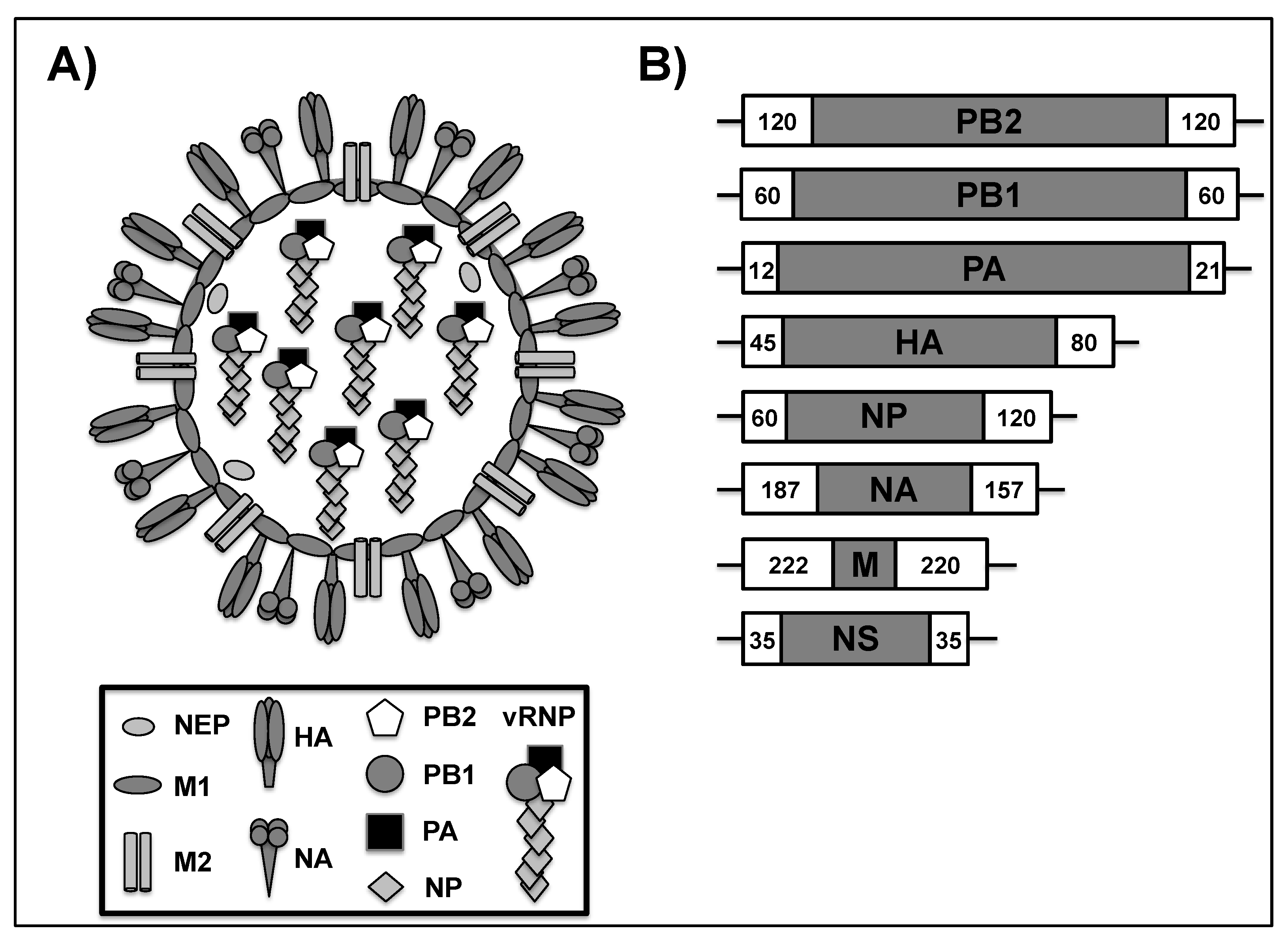
1.1. Influenza A Virus (IAV)
1.2. Current Vaccine Strategies for the Control of Human Influenza Infections
1.3. The Need of Novel LAIV Approaches to Combat Influenza Infections
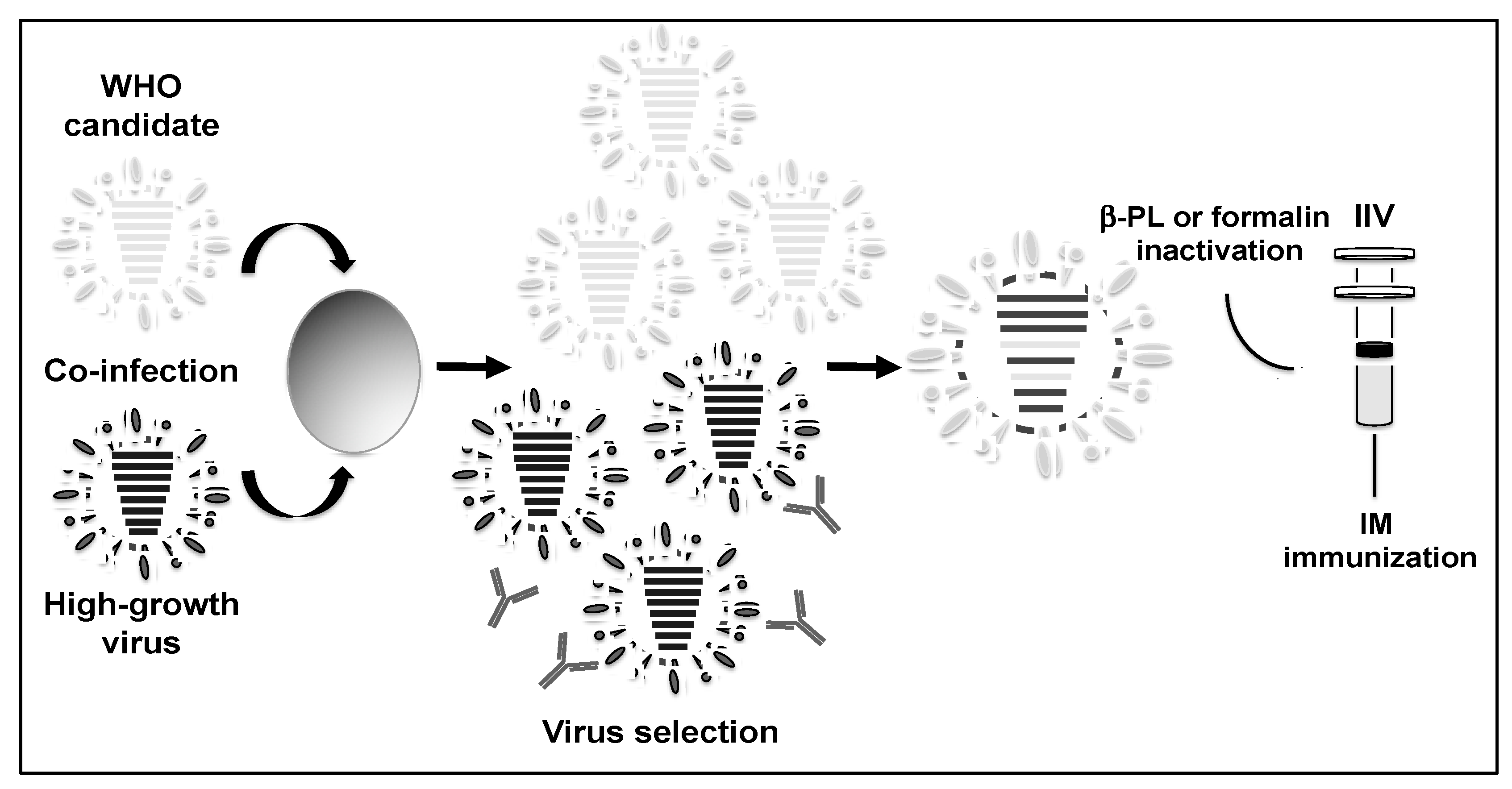
1.4. Reverse Genetics Techniques for the Development of Novel Influenza Vaccines
2. New Strategies to Develop LAIVs
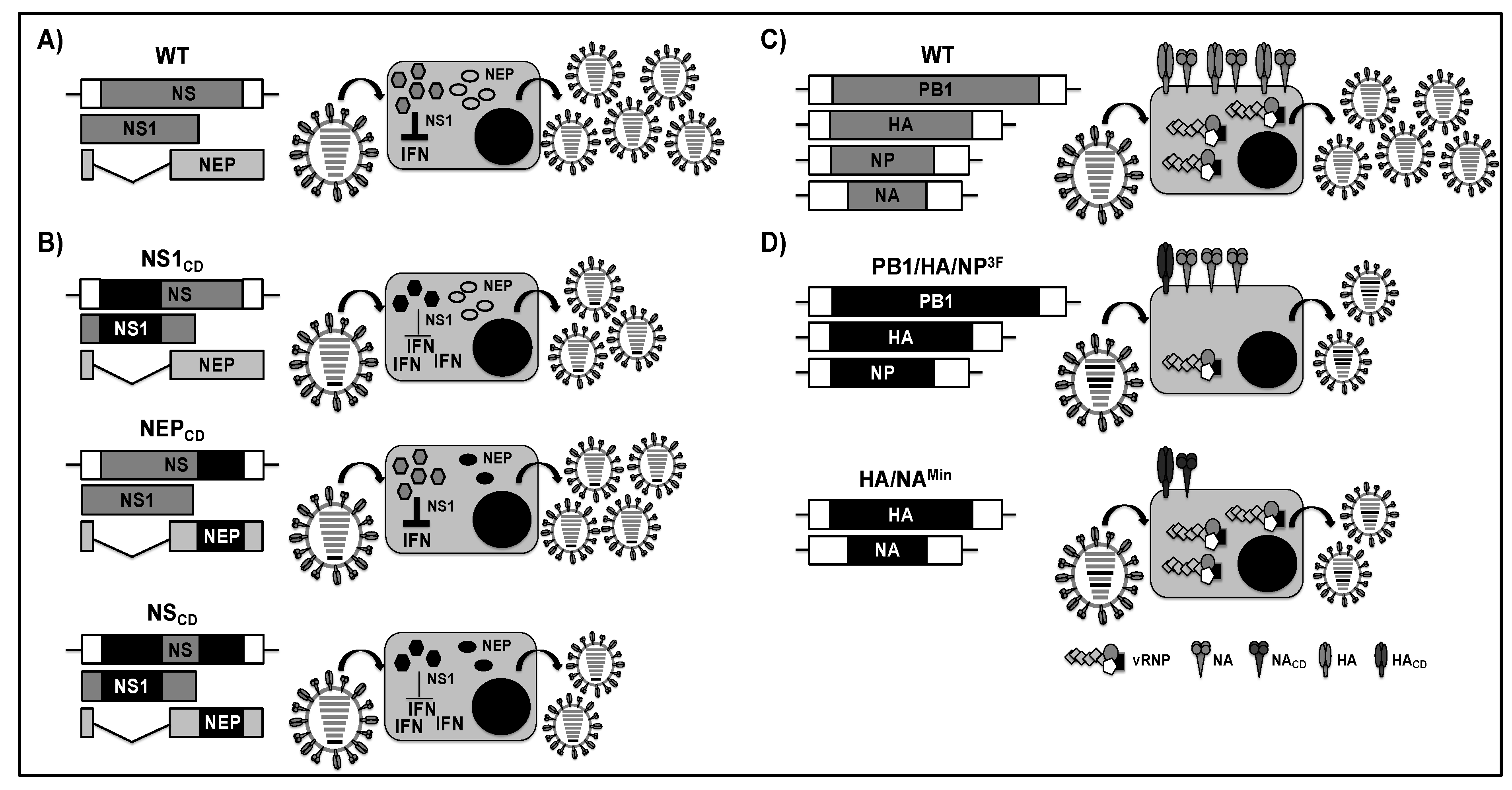
2.1. NS1 Truncated or Deficient Viruses as LAIVs
2.2. Suboptimal Codon Usage for the Development of LAIV
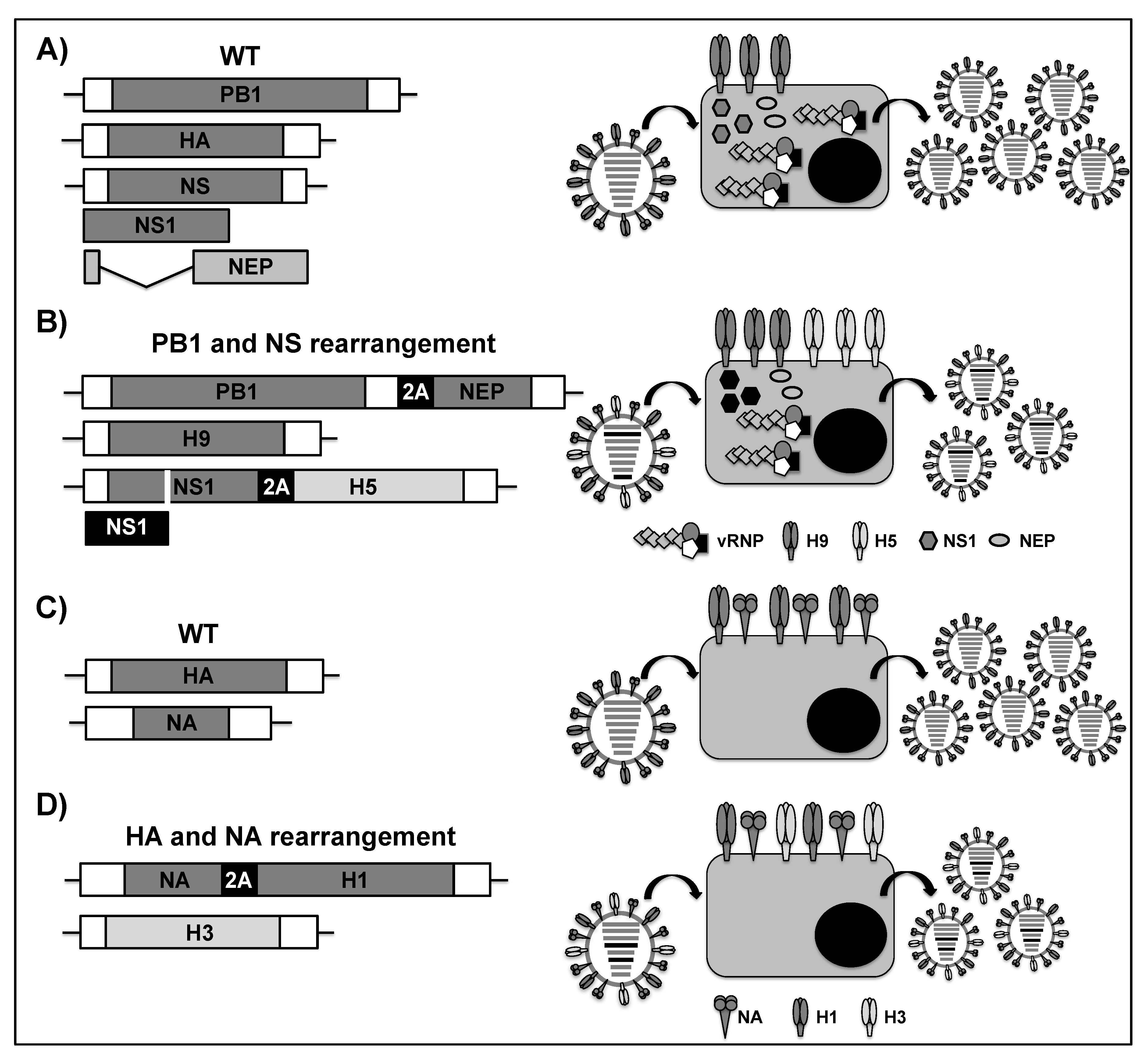
2.3. Viral Genome Rearrangement for the Development of LAIVs
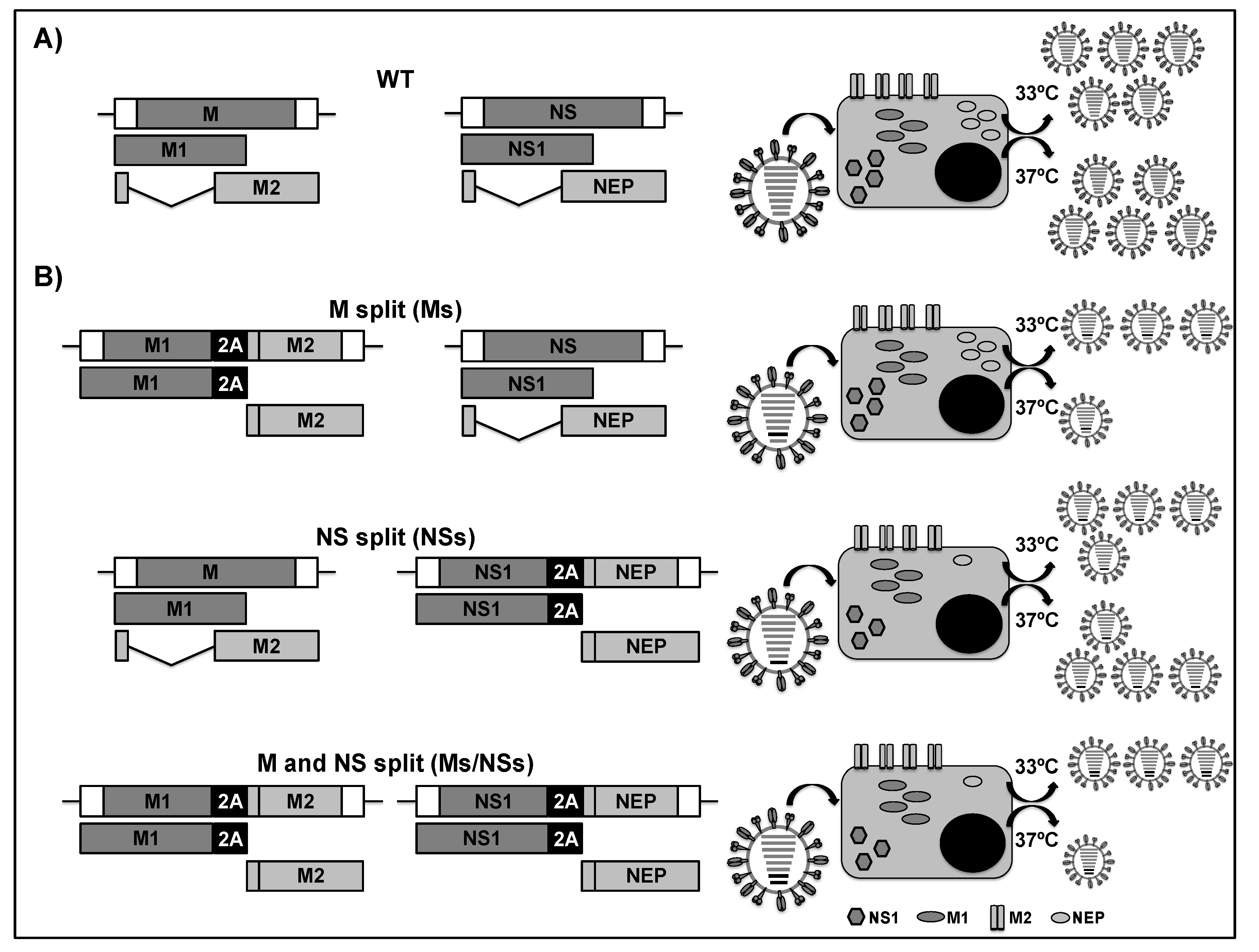
2.4. LAIVs Based on the Modification of the Viral M and NS Segments
2.5. Single-Cycle Infectious IAVs (sciIAVs) as LAIVs
2.6. LAIVs Based on Premature Termination Codon (PTC)-Harboring Viruses
3. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Shaw, M.L.; Palease, P. Orthomyxoviridae: The viruses and their replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Eds.; Lippincott Williams and WIlkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Gamblin, S.J.; Skehel, J.J. Influenza hemagglutinin and neuraminidase membrane glycoproteins. J. Biol. Chem. 2010, 285, 28403–28409. [Google Scholar] [CrossRef] [PubMed]
- Nayak, D.P.; Balogun, R.A.; Yamada, H.; Zhou, Z.H.; Barman, S. Influenza virus morphogenesis and budding. Virus Res. 2009, 143, 147–161. [Google Scholar] [CrossRef]
- Samji, T. Influenza A: Understanding the viral life cycle. Yale J. Biol. Med. 2009, 82, 153–159. [Google Scholar] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26 (Suppl. 4), D49–D53. [Google Scholar] [CrossRef]
- Neumann, G.; Brownlee, G.G.; Fodor, E.; Kawaoka, Y. Orthomyxovirus replication, transcription, and polyadenylation. Curr. Top. Microbiol. Immunol. 2004, 283, 121–143. [Google Scholar] [PubMed]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Imai, M.; Kawaoka, Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2012, 2, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, J.M.; Chan, R.W.; Russell, R.J.; Air, G.M.; Peiris, J.S. Evolving complexities of influenza virus and its receptors. Trends Microbiol. 2008, 16, 149–157. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef]
- Boivin, S.; Cusack, S.; Ruigrok, R.W.; Hart, D.J. Influenza A virus polymerase: Structural insights into replication and host adaptation mechanisms. J. Biol. Chem. 2010, 285, 28411–28417. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hutchinson, E.C.; Jagger, B.W.; Stuart, A.D.; Kang, Z.H.; Robb, N.; Schwartzman, L.M.; Kash, J.C.; Fodor, E.; Firth, A.E.; et al. Identification of a novel splice variant form of the influenza A virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012, 8, e1002998. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.; Fodor, E. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Takizawa, N.; Watanabe, K.; Nagata, K.; Kobayashi, N. Crucial role of the influenza virus NS2 (NEP) C-terminal domain in M1 binding and nuclear export of vRNP. FEBS Lett. 2011, 585, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Pohl, M.O.; Lanz, C.; Stertz, S. Late stages of the influenza A virus replication cycle—A tight interplay between virus and host. J. Gen. Virol. 2016, 97, 2058–2072. [Google Scholar] [CrossRef]
- Doms, R.W.; Lamb, R.A.; Rose, J.K.; Helenius, A. Folding and assembly of viral membrane proteins. Virology 1993, 193, 545–562. [Google Scholar] [CrossRef]
- Gerber, M.; Isel, C.; Moules, V.; Marquet, R. Selective packaging of the influenza A genome and consequences for genetic reassortment. Trends Microbiol. 2014, 22, 446–455. [Google Scholar] [CrossRef]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Colman, P.M.; Varghese, J.N.; Laver, W.G. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 1983, 303, 41–44. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Guo, Y.; Cordero, K.S.; Yu, J.; Wang, J.; Cao, Y.; Rong, L. Identification of critical residues of influenza neuraminidase in viral particle release. Virol. J. 2011, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Bridges, C.B.; Cox, N.J.; Fukuda, K. Influenza-associated hospitalizations in the United States. JAMA 2004, 292, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Fact Sheet about Seasonal Influenza; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Morris, D.E.; Cleary, D.W.; Clarke, S.C. Secondary Bacterial Infections Associated with Influenza Pandemics. Front. Microbiol. 2017, 8, 1041. [Google Scholar] [CrossRef] [PubMed]
- Morales, K.F.; Paget, J.; Spreeuwenberg, P. Possible explanations for why some countries were harder hit by the pandemic influenza virus in 2009—A global mortality impact modeling study. BMC Infect. Dis. 2017, 17, 642. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Dorratoltaj, N.; Marathe, A.; Lewis, B.L.; Swarup, S.; Eubank, S.G.; Abbas, K.M. Epidemiological and economic impact of pandemic influenza in Chicago: Priorities for vaccine interventions. PLoS Comput. Biol. 2017, 13, e1005521. [Google Scholar] [CrossRef]
- Mostafa, A.; Abdelwhab, E.M.; Mettenleiter, T.C.; Pleschka, S. Zoonotic Potential of Influenza A Viruses: A Comprehensive Overview. Viruses 2018, 10, 497. [Google Scholar] [CrossRef]
- Kassianos, G.; Blank, P.; Falup-Pecurariu, O.; Kuchar, E.; Kyncl, J.; De Lejarazu, R.O.; Nitsch-Osuch, A.; van Essen, G.A. Influenza vaccination: Key facts for general practitioners in Europe—A synthesis by European experts based on national guidelines and best practices in the United Kingdom and the Netherlands. Drugs Context 2016, 5, 212293. [Google Scholar] [CrossRef]
- Garten, R.; Blanton, L.; Elal, A.I.A.; Alabi, N.; Barnes, J.; Biggerstaff, M.; Brammer, L.; Budd, A.P.; Burns, E.; Cummings, C.N.; et al. Update: Influenza Activity in the United States During the 2017–2018 Season and Composition of the 2018–2019 Influenza Vaccine. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Sokolow, L.Z.; Broder, K.R.; Olsen, S.J.; Karron, R.A.; Jernigan, D.B.; Bresee, J.S. Prevention and Control of Seasonal Influenza with Vaccines. MMWR Recomm. Rep. 2016, 65, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Hampson, A.; Barr, I.; Cox, N.; Donis, R.O.; Siddhivinayak, H.; Jernigan, D.; Katz, J.; McCauley, J.; Motta, F.; Odagiri, T.; et al. Improving the selection and development of influenza vaccine viruses—Report of a WHO informal consultation on improving influenza vaccine virus selection, Hong Kong SAR, China, 18–20 November 2015. Vaccine 2017, 35, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Rajao, D.S.; Perez, D.R. Universal Vaccines and Vaccine Platforms to Protect against Influenza Viruses in Humans and Agriculture. Front. Microbiol. 2018, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.A.; Jones, T.C.; Barr, I.G.; Cox, N.J.; Garten, R.J.; Gregory, V.; Gust, I.D.; Hampson, A.W.; Hay, A.J.; Hurt, A.C.; et al. Influenza vaccine strain selection and recent studies on the global migration of seasonal influenza viruses. Vaccine 2008, 26 (Suppl. 4), D31–D34. [Google Scholar] [CrossRef]
- Barberis, I.; Myles, P.; Ault, S.K.; Bragazzi, N.L.; Martini, M. History and evolution of influenza control through vaccination: From the first monovalent vaccine to universal vaccines. J. Prev. Med. Hyg. 2016, 57, E115–E120. [Google Scholar]
- Miller, G.L. A Study of Conditions for the Optimum Production of Pr8 Influenza Virus in Chick Embryos. J. Exp. Med. 1944, 79, 173–183. [Google Scholar] [CrossRef]
- Wong, S.S.; Webby, R.J. Traditional and new influenza vaccines. Clin. Microbiol. Rev. 2013, 26, 476–492. [Google Scholar] [CrossRef]
- Cox, R.J.; Brokstad, K.A.; Ogra, P. Influenza virus: Immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand. J. Immunol. 2004, 59, 1–15. [Google Scholar] [CrossRef]
- Laver, W.G.; Webster, R.G. Preparation and immunogenicity of an influenza virus hemagglutinin and neuraminidase subunit vaccine. Virology 1976, 69, 511–522. [Google Scholar] [CrossRef]
- Cox, M.M.; Patriarca, P.A.; Treanor, J. FluBlok, a recombinant hemagglutinin influenza vaccine. Influenza Other Respir. Viruses 2008, 2, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Beyer, W.E.; Palache, A.M.; de Jong, J.C.; Osterhaus, A.D. Cold-adapted live influenza vaccine versus inactivated vaccine: Systemic vaccine reactions, local and systemic antibody response, and vaccine efficacy. A meta-analysis. Vaccine 2002, 20, 1340–1353. [Google Scholar] [CrossRef]
- Katayose, M.; Hosoya, M.; Haneda, T.; Yamaguchi, H.; Kawasaki, Y.; Sato, M.; Wright, P.F. The effectiveness of trivalent inactivated influenza vaccine in children over six consecutive influenza seasons. Vaccine 2011, 29, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Luksic, I.; Clay, S.; Falconer, R.; Pulanic, D.; Rudan, I.; Campbell, H.; Nair, H. Effectiveness of seasonal influenza vaccines in children—A systematic review and meta-analysis. Croat Med. J. 2013, 54, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.J.; Brokstad, K.A. The postvaccination antibody response to influenza virus proteins. APMIS 1999, 107, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Saletti, G.; Gerlach, T.; Rimmelzwaan, G.F. Influenza vaccines: ‘tailor-made’ or ‘one fits all’. Curr. Opin. Immunol. 2018, 53, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Sycheva, A.L.; Pogorelyy, M.V.; Komech, E.A.; Minervina, A.A.; Zvyagin, I.V.; Staroverov, D.B.; Chudakov, D.M.; Lebedev, Y.B.; Mamedov, I.Z. Quantitative profiling reveals minor changes of T cell receptor repertoire in response to subunit inactivated influenza vaccine. Vaccine 2018, 36, 1599–1605. [Google Scholar] [CrossRef]
- Hoft, D.F.; Lottenbach, K.R.; Blazevic, A.; Turan, A.; Blevins, T.P.; Pacatte, T.P.; Yu, Y.; Mitchell, M.C.; Hoft, S.G.; Belshe, R.B. Comparisons of the Humoral and Cellular Immune Responses Induced by Live Attenuated Influenza Vaccine and Inactivated Influenza Vaccine in Adults. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef]
- Forrest, B.D.; Pride, M.W.; Dunning, A.J.; Capeding, M.R.; Chotpitayasunondh, T.; Tam, J.S.; Rappaport, R.; Eldridge, J.H.; Gruber, W.C. Correlation of cellular immune responses with protection against culture-confirmed influenza virus in young children. Clin. Vaccine Immunol. 2008, 15, 1042–1053. [Google Scholar] [CrossRef]
- Hoft, D.F.; Babusis, E.; Worku, S.; Spencer, C.T.; Lottenbach, K.; Truscott, S.M.; Abate, G.; Sakala, I.G.; Edwards, K.M.; Creech, C.B.; et al. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J. Infect. Dis. 2011, 204, 845–853. [Google Scholar] [CrossRef]
- Mohn, K.G.; Brokstad, K.A.; Pathirana, R.D.; Bredholt, G.; Jul-Larsen, A.; Trieu, M.C.; Lartey, S.L.; Montemoli, E.; Tondel, C.; Aarstad, H.J.; et al. Live Attenuated Influenza Vaccine in Children Induces B-Cell Responses in Tonsils. J. Infect. Dis. 2016, 214, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Basha, S.; Hazenfeld, S.; Brady, R.C.; Subbramanian, R.A. Comparison of antibody and T-cell responses elicited by licensed inactivated- and live-attenuated influenza vaccines against H3N2 hemagglutinin. Hum. Immunol. 2011, 72, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Zengel, J.R.; Suguitan, A.L., Jr.; Xu, Q.; Wang, W.; Lin, J.; Jin, H. Evaluation of the humoral and cellular immune responses elicited by the live attenuated and inactivated influenza vaccines and their roles in heterologous protection in ferrets. J. Infect. Dis. 2013, 208, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lu, B.; Zhou, H.; Ma, C.; Zhao, J.; Yang, C.F.; Kemble, G.; Greenberg, H. Multiple amino acid residues confer temperature sensitivity to human influenza virus vaccine strains (FluMist) derived from cold-adapted A/Ann Arbor/6/60. Virology 2003, 306, 18–24. [Google Scholar] [CrossRef]
- Cimons, M. FDA okays live-attenuated nasal spray form of flu vaccine. Am. Soc. Microbiol. News 2003, 69, 426–427. [Google Scholar]
- Maassab, H.F. Adaptation and growth characteristics of influenza virus at 25 degrees C. Nature 1967, 213, 612–614. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.J.; Kitame, F.; Kendal, A.P.; Maassab, H.F.; Naeve, C. Identification of sequence changes in the cold-adapted, live attenuated influenza vaccine strain, A/Ann Arbor/6/60 (H2N2). Virology 1988, 167, 554–567. [Google Scholar] [CrossRef]
- Murphy, B.R.; Coelingh, K. Principles underlying the development and use of live attenuated cold-adapted influenza A and B virus vaccines. Viral Immunol. 2002, 15, 295–323. [Google Scholar] [CrossRef]
- Cox, A.; Baker, S.F.; Nogales, A.; Martinez-Sobrido, L.; Dewhurst, S. Development of a mouse-adapted live attenuated influenza virus that permits in vivo analysis of enhancements to the safety of live attenuated influenza virus vaccine. J. Virol. 2015, 89, 3421–3426. [Google Scholar] [CrossRef]
- Abramson, J.S. Intranasal, cold-adapted, live, attenuated influenza vaccine. Pediatr. Infect. Dis. J. 1999, 18, 1103–1104. [Google Scholar] [CrossRef]
- Jin, H.; Chen, Z. Production of live attenuated influenza vaccines against seasonal and potential pandemic influenza viruses. Curr. Opin. Virol. 2014, 6, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Nguyen, H.H.; Cuburu, N.; Horimoto, T.; Ko, S.Y.; Park, S.H.; Czerkinsky, C.; Kweon, M.N. Sublingual vaccination with influenza virus protects mice against lethal viral infection. Proc. Natl. Acad. Sci. USA 2008, 105, 1644–1649. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, M.R.; Souza, G.; Taishi, P.; Bohnet, S.G.; Krueger, J.M. Olfactory bulb and hypothalamic acute-phase responses to influenza virus: Effects of immunization. Neuroimmunomodulation 2013, 20, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Pronker, E.S.; Claassen, E.; Osterhaus, A.D. Development of new generation influenza vaccines: Recipes for success? Vaccine 2012, 30, 7344–7347. [Google Scholar] [CrossRef] [PubMed]
- Talbot, T.R.; Crocker, D.D.; Peters, J.; Doersam, J.K.; Ikizler, M.R.; Sannella, E.; Wright, P.E.; Edwards, K.M. Duration of virus shedding after trivalent intranasal live attenuated influenza vaccination in adults. Infect. Control Hosp. Epidemiol. 2005, 26, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Curran, M.P. Live attenuated influenza vaccine (FluMist(R); Fluenz): A review of its use in the prevention of seasonal influenza in children and adults. Drugs 2011, 71, 1591–1622. [Google Scholar] [CrossRef] [PubMed]
- Rhorer, J.; Ambrose, C.S.; Dickinson, S.; Hamilton, H.; Oleka, N.A.; Malinoski, F.J.; Wittes, J. Efficacy of live attenuated influenza vaccine in children: A meta-analysis of nine randomized clinical trials. Vaccine 2009, 27, 1101–1110. [Google Scholar] [CrossRef]
- Graaf, H.; Faust, S.N. Fluarix quadrivalent vaccine for influenza. Expert Rev. Vaccines 2015, 14, 1055–1063. [Google Scholar] [CrossRef]
- Chen, Z.; Aspelund, A.; Kemble, G.; Jin, H. Molecular studies of temperature-sensitive replication of the cold-adapted B/Ann Arbor/1/66, the master donor virus for live attenuated influenza FluMist vaccines. Virology 2008, 380, 354–362. [Google Scholar] [CrossRef]
- Fiore, A.E.; Uyeki, T.M.; Broder, K.; Finelli, L.; Euler, G.L.; Singleton, J.A.; Iskander, J.K.; Wortley, P.M.; Shay, D.K.; Bresee, J.S.; et al. Prevention and control of influenza with vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recomm. Rep. 2010, 59, 1–62. [Google Scholar]
- Greenhawt, M.J. Influenza vaccination in asthmatic patients. J. Allergy Clin. Immunol. 2014, 133, 1233–1234. [Google Scholar] [CrossRef] [PubMed]
- Lewnard, J.A.; Cobey, S. Immune History and Influenza Vaccine Effectiveness. Vaccines 2018, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Manzoli, L.; Ioannidis, J.P.; Flacco, M.E.; De Vito, C.; Villari, P. Effectiveness and harms of seasonal and pandemic influenza vaccines in children, adults and elderly: A critical review and re-analysis of 15 meta-analyses. Hum. Vaccines Immunother. 2012, 8, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Cox, N.J. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 1990, 8, 737–771. [Google Scholar] [CrossRef] [PubMed]
- Singanayagam, A.; Zambon, M.; Lalvani, A.; Barclay, W. Urgent challenges in implementing live attenuated influenza vaccine. Lancet Infect. Dis. 2018, 18, e25–e32. [Google Scholar] [CrossRef]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Ambrose, C.S.; Levin, M.J.; Belshe, R.B. The relative efficacy of trivalent live attenuated and inactivated influenza vaccines in children and adults. Influenza Other. Respir. Viruses 2011, 5, 67–75. [Google Scholar] [CrossRef]
- Jefferson, T.; Rivetti, A.; Di Pietrantonj, C.; Demicheli, V.; Ferroni, E. Vaccines for preventing influenza in healthy children. Cochrane Database Syst. Rev. 2008, CD004879. [Google Scholar] [CrossRef]
- Chung, J.R.; Flannery, B.; Thompson, M.G.; Gaglani, M.; Jackson, M.L.; Monto, A.S.; Nowalk, M.P.; Talbot, H.K.; Treanor, J.J.; Belongia, E.A.; et al. Seasonal Effectiveness of Live Attenuated and Inactivated Influenza Vaccine. Pediatrics 2016, 137, e20153279. [Google Scholar] [CrossRef]
- McLean, H.Q.; Caspard, H.; Griffin, M.R.; Poehling, K.A.; Gaglani, M.; Belongia, E.A.; Talbot, H.K.; Peters, T.R.; Murthy, K.; Ambrose, C.S. Effectiveness of live attenuated influenza vaccine and inactivated influenza vaccine in children during the 2014–2015 season. Vaccine 2017, 35, 2685–2693. [Google Scholar] [CrossRef]
- Poehling, K.A.; Caspard, H.; Peters, T.R.; Belongia, E.A.; Congeni, B.; Gaglani, M.; Griffin, M.R.; Irving, S.A.; Kavathekar, P.K.; McLean, H.Q.; et al. 2015-2016 Vaccine Effectiveness of Live Attenuated and Inactivated Influenza Vaccines in Children in the United States. Clin. Infect. Dis. 2018, 66, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, D.M.; Chambers, C.; Sabaiduc, S.; De Serres, G.; Winter, A.L.; Dickinson, J.A.; Gubbay, J.B.; Drews, S.J.; Martineau, C.; Charest, H.; et al. Beyond Antigenic Match: Possible Agent-Host and Immuno-epidemiological Influences on Influenza Vaccine Effectiveness During the 2015–2016 Season in Canada. J. Infect. Dis. 2017, 216, 1487–1500. [Google Scholar] [CrossRef] [PubMed]
- Caspard, H.; Mallory, R.M.; Yu, J.; Ambrose, C.S. Live-Attenuated Influenza Vaccine Effectiveness in Children From 2009 to 2015-2016: A Systematic Review and Meta-Analysis. Open Forum Infect. Dis. 2017, 4, ofx111. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Sokolow, L.Z.; Broder, K.R.; Walter, E.B.; Bresee, J.S.; Fry, A.M.; Jernigan, D.B. Prevention and Control of Seasonal Influenza With Vaccines: Recommendations of the Advisory Committee on Immunization Practices-United States, 2017-18 Influenza Season. Am. J. Transpl. 2017, 17, 2970–2982. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Sokolow, L.Z.; Fry, A.M.; Walter, E.B.; Jernigan, D.B. Update: ACIP Recommendations for the Use of Quadrivalent Live Attenuated Influenza Vaccine (LAIV4)—United States, 2018–19 Influenza Season. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 643–645. [Google Scholar] [CrossRef] [PubMed]
- Pebody, R.; McMenamin, J.; Nohynek, H. Live attenuated influenza vaccine (LAIV): Recent effectiveness results from the USA and implications for LAIV programmes elsewhere. Arch. Dis. Child. 2018, 103, 101–105. [Google Scholar] [CrossRef]
- Ambrose, C.S.; Bright, H.; Mallory, R. Letter to the editor: Potential causes of the decreased effectiveness of the influenza A(H1N1)pdm09 strain in live attenuated influenza vaccines. Eurosurveill 2016, 21, 30394. [Google Scholar] [CrossRef]
- Sridhar, S.; Begom, S.; Bermingham, A.; Hoschler, K.; Adamson, W.; Carman, W.; Bean, T.; Barclay, W.; Deeks, J.J.; Lalvani, A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat. Med. 2013, 19, 1305–1312. [Google Scholar] [CrossRef]
- Elderfield, R.A.; Watson, S.J.; Godlee, A.; Adamson, W.E.; Thompson, C.I.; Dunning, J.; Fernandez-Alonso, M.; Blumenkrantz, D.; Hussell, T.; Investigators, M.; et al. Accumulation of human-adapting mutations during circulation of A(H1N1)pdm09 influenza virus in humans in the United Kingdom. J. Virol. 2014, 88, 13269–13283. [Google Scholar] [CrossRef]
- Russier, M.; Yang, G.; Rehg, J.E.; Wong, S.S.; Mostafa, H.H.; Fabrizio, T.P.; Barman, S.; Krauss, S.; Webster, R.G.; Webby, R.J.; et al. Molecular requirements for a pandemic influenza virus: An acid-stable hemagglutinin protein. Proc. Natl. Acad. Sci. USA 2016, 113, 1636–1641. [Google Scholar] [CrossRef]
- O’Donnell, C.D.; Vogel, L.; Matsuoka, Y.; Jin, H.; Subbarao, K. The matrix gene segment destabilizes the acid and thermal stability of the hemagglutinin of pandemic live attenuated influenza virus vaccines. J. Virol. 2014, 88, 12374–12384. [Google Scholar] [CrossRef] [PubMed]
- Laurie, K.L.; Guarnaccia, T.A.; Carolan, L.A.; Yan, A.W.; Aban, M.; Petrie, S.; Cao, P.; Heffernan, J.M.; McVernon, J.; Mosse, J.; et al. Interval Between Infections and Viral Hierarchy Are Determinants of Viral Interference Following Influenza Virus Infection in a Ferret Model. J. Infect. Dis. 2015, 212, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Caspard, H.; Coelingh, K.L.; Mallory, R.M.; Ambrose, C.S. Association of vaccine handling conditions with effectiveness of live attenuated influenza vaccine against H1N1pdm09 viruses in the United States. Vaccine 2016, 34, 5066–5072. [Google Scholar] [CrossRef] [PubMed]
- Gould, P.S.; Easton, A.J.; Dimmock, N.J. Live Attenuated Influenza Vaccine contains Substantial and Unexpected Amounts of Defective Viral Genomic RNA. Viruses 2017, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Kroger, A.T.; Atkinson, W.L.; Marcuse, E.K.; Pickering, L.K.; Advisory Committee on Immunization Practices (ACIP) Centers for Disease Control and Prevention (CDC). General recommendations on immunization: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm. Rep. 2011, 60, 1–64. [Google Scholar]
- Sridhar, S.; Brokstad, K.A.; Cox, R.J. Influenza Vaccination Strategies: Comparing Inactivated and Live Attenuated Influenza Vaccines. Vaccines 2015, 3, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Rodriguez, L.; Chauche, C.; Huang, K.; Reilly, E.C.; Topham, D.J.; Murcia, P.R.; Parrish, C.R.; Martinez-Sobrido, L. Temperature-Sensitive Live-Attenuated Canine Influenza Virus H3N8 Vaccine. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Friede, M.; Palkonyay, L.; Alfonso, C.; Pervikov, Y.; Torelli, G.; Wood, D.; Kieny, M.P. WHO initiative to increase global and equitable access to influenza vaccine in the event of a pandemic: Supporting developing country production capacity through technology transfer. Vaccine 2011, 29 (Suppl. 1), A2–A7. [Google Scholar] [CrossRef]
- Burgess, T.H.; Murray, C.K.; Bavaro, M.F.; Landrum, M.L.; O’Bryan, T.A.; Rosas, J.G.; Cammarata, S.M.; Martin, N.J.; Ewing, D.; Raviprakash, K.; et al. Self-administration of intranasal influenza vaccine: Immunogenicity and volunteer acceptance. Vaccine 2015, 33, 3894–3899. [Google Scholar] [CrossRef]
- Baker, S.F.; Nogales, A.; Finch, C.; Tuffy, K.M.; Domm, W.; Perez, D.R.; Topham, D.J.; Martinez-Sobrido, L. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J. Virol. 2014, 88, 10778–10791. [Google Scholar] [CrossRef]
- Jackson, D.; Elderfield, R.A.; Barclay, W.S. Molecular studies of influenza B virus in the reverse genetics era. J. Gen. Virol. 2011, 92, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sobrido, L.; Garcia-Sastre, A. Generation of recombinant influenza virus from plasmid DNA. J. Vis. Exp. 2010. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.G. Many ways to make an influenza virus—Review of influenza virus reverse genetics methods. Influenza Other. Respir. Viruses 2013, 7, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sobrido, L.; Cadagan, R.; Steel, J.; Basler, C.F.; Palese, P.; Moran, T.M.; Garcia-Sastre, A. Hemagglutinin-pseudotyped green fluorescent protein-expressing influenza viruses for the detection of influenza virus neutralizing antibodies. J. Virol. 2010, 84, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Domm, W.; Martinez-Sobrido, L. Development and applications of single-cycle infectious influenza A virus (sciIAV). Virus Res. 2016, 216, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Martinez-Sobrido, L. Replication-competent influenza A viruses expressing a red fluorescent protein. Virology 2015, 476, 206–216. [Google Scholar] [CrossRef]
- Breen, M.; Nogales, A.; Baker, S.F.; Martinez-Sobrido, L. Replication-Competent Influenza A Viruses Expressing Reporter Genes. Viruses 2016, 8, 179. [Google Scholar] [CrossRef]
- Breen, M.; Nogales, A.; Baker, S.F.; Perez, D.R.; Martinez-Sobrido, L. Replication-Competent Influenza A and B Viruses Expressing a Fluorescent Dynamic Timer Protein for In Vitro and In Vivo Studies. PLoS ONE 2016, 11, e0147723. [Google Scholar] [CrossRef]
- Rodriguez, L.; Nogales, A.; Murcia, P.R.; Parrish, C.R.; Martinez-Sobrido, L. A bivalent live-attenuated influenza vaccine for the control and prevention of H3N8 and H3N2 canine influenza viruses. Vaccine 2017, 35, 4374–4381. [Google Scholar] [CrossRef]
- Rodriguez, L.; Nogales, A.; Reilly, E.C.; Topham, D.J.; Murcia, P.R.; Parrish, C.R.; Martinez Sobrido, L. A live-attenuated influenza vaccine for H3N2 canine influenza virus. Virology 2017, 504, 96–106. [Google Scholar] [CrossRef]
- Rodriguez, L.; Reedy, S.; Nogales, A.; Murcia, P.R.; Chambers, T.M.; Martinez-Sobrido, L. Development of a novel equine influenza virus live-attenuated vaccine. Virology 2018, 516, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L. Reverse Genetics Approaches for the Development of Influenza Vaccines. Int. J. Mol. Sci. 2016, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Kawaoka, Y.; Hobom, G.; Webster, R.G. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. USA 2000, 97, 6108–6113. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Neumann, G.; Hobom, G.; Webster, R.G.; Kawaoka, Y. “Ambisense” approach for the generation of influenza A virus: vRNA and mRNA synthesis from one template. Virology 2000, 267, 310–317. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). WHO Guidance on Development of Influenza Vaccine Reference Viruses by Reverse Genetics; WHO: Geneva, Switzerland, 2005. [Google Scholar]
- Perdue, M.L.; Arnold, F.; Li, S.; Donabedian, A.; Cioce, V.; Warf, T.; Huebner, R. The future of cell culture-based influenza vaccine production. Expert Rev. Vaccines 2011, 10, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Hegde, N.R. Cell culture-based influenza vaccines: A necessary and indispensable investment for the future. Hum. Vaccines Immunother. 2015, 11, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Trombetta, C.M.; Lazzeri, G.; Pozzi, T.; Rossi, S.; Montomoli, E. Egg-Independent Influenza Vaccines and Vaccine Candidates. Vaccines 2017, 5, 18. [Google Scholar] [CrossRef]
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2009, 8, 607–618. [Google Scholar] [CrossRef]
- Vlecken, D.H.; Pelgrim, R.P.; Ruminski, S.; Bakker, W.A.; van der Pol, L.A. Comparison of initial feasibility of host cell lines for viral vaccine production. J. Virol. Methods 2013, 193, 28–41. [Google Scholar] [CrossRef]
- Harding, A.T.; Heaton, N.S. Efforts to Improve the Seasonal Influenza Vaccine. Vaccines 2018, 6, 19. [Google Scholar] [CrossRef]
- Barr, I.G.; Donis, R.O.; Katz, J.M.; McCauley, J.W.; Odagiri, T.; Trusheim, H.; Tsai, T.F.; Wentworth, D.E. Cell culture-derived influenza vaccines in the severe 2017–2018 epidemic season: A step towards improved influenza vaccine effectiveness. NPJ Vaccines 2018, 3, 44. [Google Scholar] [CrossRef] [PubMed]
- Fulvini, A.A.; Ramanunninair, M.; Le, J.; Pokorny, B.A.; Arroyo, J.M.; Silverman, J.; Devis, R.; Bucher, D. Gene constellation of influenza A virus reassortants with high growth phenotype prepared as seed candidates for vaccine production. PLoS ONE 2011, 6, e20823. [Google Scholar] [CrossRef]
- Del Giudice, G.; Rappuoli, R. Inactivated and adjuvanted influenza vaccines. Curr. Top. Microbiol. Immunol. 2015, 386, 151–180. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, C.; Major, D.; Wood, J.M.; Robertson, J.S. Generation of influenza vaccine viruses on Vero cells by reverse genetics: An H5N1 candidate vaccine strain produced under a quality system. Vaccine 2005, 23, 2943–2952. [Google Scholar] [CrossRef]
- Robertson, J.S.; Engelhardt, O.G. Developing vaccines to combat pandemic influenza. Viruses 2010, 2, 532–546. [Google Scholar] [CrossRef]
- Marill, M.C. After flu vaccine mismatch, calls for delayed selection intensify. Nat. Med. 2015, 21, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Tisoncik, J.R.; Billharz, R.; Burmakina, S.; Belisle, S.E.; Proll, S.C.; Korth, M.J.; Garcia-Sastre, A.; Katze, M.G. The NS1 protein of influenza A virus suppresses interferon-regulated activation of antigen-presentation and immune-proteasome pathways. J. Gen. Virol. 2011, 92, 2093–2104. [Google Scholar] [CrossRef]
- Thulasi Raman, S.N.; Zhou, Y. Networks of Host Factors that Interact with NS1 Protein of Influenza A Virus. Front. Microbiol. 2016, 7, 654. [Google Scholar] [CrossRef]
- Hsu, A.C. Influenza Virus: A Master Tactician in Innate Immune Evasion and Novel Therapeutic Interventions. Front. Immunol. 2018, 9, 743. [Google Scholar] [CrossRef]
- Chauche, C.; Nogales, A.; Zhu, H.; Goldfarb, D.; Ahmad Shanizza, A.I.; Gu, Q.; Parrish, C.R.; Martinez-Sobrido, L.; Marshall, J.F.; Murcia, P.R. Mammalian Adaptation of an Avian Influenza A Virus Involves Stepwise Changes in NS1. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Functional Evolution of Influenza Virus NS1 Protein in Currently Circulating Human 2009 Pandemic H1N1 Viruses. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- DeDiego, M.L.; Nogales, A.; Lambert-Emo, K.; Martinez-Sobrido, L.; Topham, D.J. NS1 Protein Mutation I64T Affects Interferon Responses and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2016, 90, 9693–9711. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Chauche, C.; DeDiego, M.L.; Topham, D.J.; Parrish, C.R.; Murcia, P.R.; Martinez-Sobrido, L. The K186E Amino Acid Substitution in the Canine Influenza Virus H3N8 NS1 Protein Restores Its Ability to Inhibit Host Gene Expression. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Huang, K.; Chauche, C.; DeDiego, M.L.; Murcia, P.R.; Parrish, C.R.; Martinez-Sobrido, L. Canine influenza viruses with modified NS1 proteins for the development of live-attenuated vaccines. Virology 2017, 500, 1–10. [Google Scholar] [CrossRef]
- Nogales, A.; Martinez-Sobrido, L.; Chiem, K.; Topham, D.J.; DeDiego, M.L. Functional Evolution of the 2009 Pandemic H1n1 Influenza Ns1 and Pa in Humans. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. NS1 Protein Amino Acid Changes D189N and V194I Affect Interferon Responses, Thermosensitivity, and Virulence of Circulating H3N2 Human Influenza A Viruses. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Nogales, A.; Rodriguez, L.; DeDiego, M.L.; Topham, D.J.; Martinez-Sobrido, L. Interplay of PA-X and NS1 Proteins in Replication and Pathogenesis of a Temperature-Sensitive 2009 Pandemic H1N1 Influenza A Virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- García-Sastre, A.; Egorov, A.; Matassov, D.; Brandt, S.; Levy, D.E.; Durbin, J.E.; Palese, P.; Muster, T. Influenza A virus lacking the NS1 gene replicates in interferon-deficient systems. Virology 1998, 252, 324–330. [Google Scholar] [CrossRef]
- Falcon, A.M.; Fernandez-Sesma, A.; Nakaya, Y.; Moran, T.M.; Ortin, J.; Garcia-Sastre, A. Attenuation and immunogenicity in mice of temperature-sensitive influenza viruses expressing truncated NS1 proteins. J. Gen. Virol. 2005, 86, 2817–2821. [Google Scholar] [CrossRef]
- Chen, C.; Fan, W.; Li, J.; Zheng, W.; Zhang, S.; Yang, L.; Liu, D.; Liu, W.; Sun, L. A Promising IFN-Deficient System to Manufacture IFN-Sensitive Influenza Vaccine Virus. Front. Cell. Infect. Microbiol. 2018, 8, 127. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Choppin, P.W. Segment 8 of the influenza virus genome is unique in coding for two polypeptides. Proc. Natl. Acad. Sci. USA 1979, 76, 4908–4912. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, M.; Zamarin, D.; Garcia-Sastre, A.; Cullinane, A.; Chambers, T.; Palese, P. Attenuation of equine influenza viruses through truncations of the NS1 protein. J. Virol. 2005, 79, 8431–8439. [Google Scholar] [CrossRef] [PubMed]
- Richt, J.A.; Lekcharoensuk, P.; Lager, K.M.; Vincent, A.L.; Loiacono, C.M.; Janke, B.H.; Wu, W.H.; Yoon, K.J.; Webby, R.J.; Solorzano, A.; et al. Vaccination of pigs against swine influenza viruses by using an NS1-truncated modified live-virus vaccine. J. Virol. 2006, 80, 11009–11018. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, A.; Webby, R.J.; Lager, K.M.; Janke, B.H.; Garcia-Sastre, A.; Richt, J.A. Mutations in the NS1 protein of swine influenza virus impair anti-interferon activity and confer attenuation in pigs. J. Virol. 2005, 79, 7535–7543. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.L.; Ma, W.; Lager, K.M.; Janke, B.H.; Webby, R.J.; Garcia-Sastre, A.; Richt, J.A. Efficacy of intranasal administration of a truncated NS1 modified live influenza virus vaccine in swine. Vaccine 2007, 25, 7999–8009. [Google Scholar] [CrossRef]
- Kappes, M.A.; Sandbulte, M.R.; Platt, R.; Wang, C.; Lager, K.M.; Henningson, J.N.; Lorusso, A.; Vincent, A.L.; Loving, C.L.; Roth, J.A.; et al. Vaccination with NS1-truncated H3N2 swine influenza virus primes T cells and confers cross-protection against an H1N1 heterosubtypic challenge in pigs. Vaccine 2012, 30, 280–288. [Google Scholar] [CrossRef]
- Steel, J.; Lowen, A.C.; Pena, L.; Angel, M.; Solorzano, A.; Albrecht, R.; Perez, D.R.; Garcia-Sastre, A.; Palese, P. Live attenuated influenza viruses containing NS1 truncations as vaccine candidates against H5N1 highly pathogenic avian influenza. J. Virol. 2009, 83, 1742–1753. [Google Scholar] [CrossRef]
- Wang, L.; Suarez, D.L.; Pantin-Jackwood, M.; Mibayashi, M.; Garcia-Sastre, A.; Saif, Y.M.; Lee, C.W. Characterization of influenza virus variants with different sizes of the non-structural (NS) genes and their potential as a live influenza vaccine in poultry. Vaccine 2008, 26, 3580–3586. [Google Scholar] [CrossRef]
- Choi, E.H.; Song, M.S.; Park, S.J.; Pascua, P.N.; Baek, Y.H.; Kwon, H.I.; Kim, E.H.; Kim, S.; Jang, H.K.; Poo, H.; et al. Development of a dual-protective live attenuated vaccine against H5N1 and H9N2 avian influenza viruses by modifying the NS1 gene. Arch. Virol. 2015, 160, 1729–1740. [Google Scholar] [CrossRef]
- Jang, H.; Ngunjiri, J.M.; Lee, C.W. Association between Interferon Response and Protective Efficacy of NS1-Truncated Mutants as Influenza Vaccine Candidates in Chickens. PLoS ONE 2016, 11, e0156603. [Google Scholar] [CrossRef]
- Jang, H.; Elaish, M.; Kc, M.; Abundo, M.C.; Ghorbani, A.; Ngunjiri, J.M.; Lee, C.W. Efficacy and synergy of live-attenuated and inactivated influenza vaccines in young chickens. PLoS ONE 2018, 13, e0195285. [Google Scholar] [CrossRef] [PubMed]
- Baskin, C.R.; Bielefeldt-Ohmann, H.; Garcia-Sastre, A.; Tumpey, T.M.; Van Hoeven, N.; Carter, V.S.; Thomas, M.J.; Proll, S.; Solorzano, A.; Billharz, R.; et al. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. J. Virol. 2007, 81, 11817–11827. [Google Scholar] [CrossRef] [PubMed]
- Pica, N.; Langlois, R.A.; Krammer, F.; Margine, I.; Palese, P. NS1-truncated live attenuated virus vaccine provides robust protection to aged mice from viral challenge. J. Virol. 2012, 86, 10293–10301. [Google Scholar] [CrossRef]
- Wacheck, V.; Egorov, A.; Groiss, F.; Pfeiffer, A.; Fuereder, T.; Hoeflmayer, D.; Kundi, M.; Popow-Kraupp, T.; Redlberger-Fritz, M.; Mueller, C.A.; et al. A novel type of influenza vaccine: Safety and immunogenicity of replication-deficient influenza virus created by deletion of the interferon antagonist NS1. J. Infect. Dis. 2010, 201, 354–362. [Google Scholar] [CrossRef]
- Talon, J.; Salvatore, M.; O’Neill, R.E.; Nakaya, Y.; Zheng, H.; Muster, T.; Garcia-Sastre, A.; Palese, P. Influenza A and B viruses expressing altered NS1 proteins: A vaccine approach. Proc. Natl. Acad. Sci. USA 2000, 97, 4309–4314. [Google Scholar] [CrossRef] [PubMed]
- Mossler, C.; Groiss, F.; Wolzt, M.; Wolschek, M.; Seipelt, J.; Muster, T. Phase I/II trial of a replication-deficient trivalent influenza virus vaccine lacking NS1. Vaccine 2013, 31, 6194–6200. [Google Scholar] [CrossRef]
- Wang, X.; Basler, C.F.; Williams, B.R.; Silverman, R.H.; Palese, P.; Garcia-Sastre, A. Functional replacement of the carboxy-terminal two-thirds of the influenza A virus NS1 protein with short heterologous dimerization domains. J. Virol. 2002, 76, 12951–12962. [Google Scholar] [CrossRef]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes and consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Biro, J.C. Does codon bias have an evolutionary origin? Biol. Med. Model. 2008, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Bera, B.C.; Greenbaum, B.D.; Bhatia, S.; Sood, R.; Selvaraj, P.; Anand, T.; Tripathi, B.N.; Virmani, N. Revelation of Influencing Factors in Overall Codon Usage Bias of Equine Influenza Viruses. PLoS ONE 2016, 11, e0154376. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef]
- Goni, N.; Iriarte, A.; Comas, V.; Sonora, M.; Moreno, P.; Moratorio, G.; Musto, H.; Cristina, J. Pandemic influenza A virus codon usage revisited: Biases, adaptation and implications for vaccine strain development. Virol. J. 2012, 9, 263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.P.; Ying, D.Q.; Li, P.; Li, F.; Bo, X.C.; Wang, S.Q. Analysis of synonymous codon usage bias in 09H1N1. Virol. Sin. 2010, 25, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Gu, W.; Ma, J.; Sun, X.; Lu, Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems 2005, 81, 77–86. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Mueller, S.; Coleman, J.R.; Papamichail, D.; Ward, C.B.; Nimnual, A.; Futcher, B.; Skiena, S.; Wimmer, E. Live attenuated influenza virus vaccines by computer-aided rational design. Nat. Biotechnol. 2010, 28, 723–726. [Google Scholar] [CrossRef]
- Yang, C.; Skiena, S.; Futcher, B.; Mueller, S.; Wimmer, E. Deliberate reduction of hemagglutinin and neuraminidase expression of influenza virus leads to an ultraprotective live vaccine in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 9481–9486. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Nogales, A.; Martinez-Sobrido, L. Downregulating viral gene expression: Codon usage bias manipulation for the generation of novel influenza A virus vaccines. Future Virol. 2015, 10, 715–730. [Google Scholar] [CrossRef]
- Fan, R.L.; Valkenburg, S.A.; Wong, C.K.; Li, O.T.; Nicholls, J.M.; Rabadan, R.; Peiris, J.S.; Poon, L.L. Generation of Live Attenuated Influenza Virus by Using Codon Usage Bias. J. Virol. 2015, 89, 10762–10773. [Google Scholar] [CrossRef] [PubMed]
- Broadbent, A.J.; Santos, C.P.; Anafu, A.; Wimmer, E.; Mueller, S.; Subbarao, K. Evaluation of the attenuation, immunogenicity, and efficacy of a live virus vaccine generated by codon-pair bias de-optimization of the 2009 pandemic H1N1 influenza virus, in ferrets. Vaccine 2016, 34, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Baker, S.F.; Ortiz-Riano, E.; Dewhurst, S.; Topham, D.J.; Martinez-Sobrido, L. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J. Virol. 2014, 88, 10525–10540. [Google Scholar] [CrossRef] [PubMed]
- Jack, B.R.; Boutz, D.R.; Paff, M.L.; Smith, B.L.; Bull, J.J.; Wilke, C.O. Reduced Protein Expression in a Virus Attenuated by Codon Deoptimization. G3 (Bethesda) 2017, 7, 2957–2968. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Papamichail, D.; Coleman, J.R.; Skiena, S.; Wimmer, E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J. Virol. 2006, 80, 9687–9696. [Google Scholar] [CrossRef] [PubMed]
- Sutejo, R.; Yeo, D.S.; Myaing, M.Z.; Hui, C.; Xia, J.; Ko, D.; Cheung, P.C.; Tan, B.H.; Sugrue, R.J. Activation of type I and III interferon signalling pathways occurs in lung epithelial cells infected with low pathogenic avian influenza viruses. PLoS ONE 2012, 7, e33732. [Google Scholar] [CrossRef]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef]
- Moura, G.; Pinheiro, M.; Arrais, J.; Gomes, A.C.; Carreto, L.; Freitas, A.; Oliveira, J.L.; Santos, M.A. Large scale comparative codon-pair context analysis unveils general rules that fine-tune evolution of mRNA primary structure. PLoS ONE 2007, 2, e847. [Google Scholar] [CrossRef]
- Biggerstaff, M.; Cauchemez, S.; Reed, C.; Gambhir, M.; Finelli, L. Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: A systematic review of the literature. BMC Infect. Dis. 2014, 14, 480. [Google Scholar] [CrossRef]
- Parvin, R.; Begum, J.A.; Nooruzzaman, M.; Chowdhury, E.H.; Islam, M.R.; Vahlenkamp, T.W. Review analysis and impact of co-circulating H5N1 and H9N2 avian influenza viruses in Bangladesh. Epidemiol. Infect. 2018, 146, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Pena, L.; Sutton, T.; Chockalingam, A.; Kumar, S.; Angel, M.; Shao, H.; Chen, H.; Li, W.; Perez, D.R. Influenza viruses with rearranged genomes as live-attenuated vaccines. J. Virol. 2013, 87, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.T.; Heaton, B.E.; Dumm, R.E.; Heaton, N.S. Rationally Designed Influenza Virus Vaccines That Are Antigenically Stable during Growth in Eggs. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lu, B.; Zhou, H.; Ye, D.; Kemble, G.; Jin, H. Improvement of influenza A/Fujian/411/02 (H3N2) virus growth in embryonated chicken eggs by balancing the hemagglutinin and neuraminidase activities, using reverse genetics. J. Virol. 2005, 79, 6763–6771. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; DeDiego, M.L.; Topham, D.J.; Martinez-Sobrido, L. Rearrangement of Influenza Virus Spliced Segments for the Development of Live-Attenuated Vaccines. J. Virol. 2016, 90, 6291–6302. [Google Scholar] [CrossRef]
- Dudek, T.; Knipe, D.M. Replication-defective viruses as vaccines and vaccine vectors. Virology 2006, 344, 230–239. [Google Scholar] [CrossRef]
- Bussow, K. Stable mammalian producer cell lines for structural biology. Curr. Opin. Struct. Biol. 2015, 32, 81–90. [Google Scholar] [CrossRef]
- Lanza, A.M.; Kim, D.S.; Alper, H.S. Evaluating the influence of selection markers on obtaining selected pools and stable cell lines in human cells. Biotechnol. J. 2013, 8, 811–821. [Google Scholar] [CrossRef]
- Shih, F.F.; Cerasoli, D.M.; Caton, A.J. A major T cell determinant from the influenza virus hemagglutinin (HA) can be a cryptic self peptide in HA transgenic mice. Int. Immunol. 1997, 9, 249–261. [Google Scholar] [CrossRef]
- Inagaki, A.; Goto, H.; Kakugawa, S.; Ozawa, M.; Kawaoka, Y. Competitive incorporation of homologous gene segments of influenza A virus into virions. J. Virol. 2012, 86, 10200–10202. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Hatami, R.; Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007, 81, 9727–9736. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Victor, S.T.; Taft, A.S.; Yamada, S.; Li, C.; Hatta, M.; Das, S.C.; Takashita, E.; Kakugawa, S.; Maher, E.A.; et al. Replication-incompetent influenza A viruses that stably express a foreign gene. J. Gen. Virol. 2011, 92, 2879–2888. [Google Scholar] [CrossRef] [PubMed]
- Bottini, A.; De, S.K.; Baaten, B.J.; Wu, B.; Barile, E.; Soonthornvacharin, S.; Stebbins, J.L.; Bradley, L.M.; Chanda, S.K.; Pellecchia, M. Identification of small molecules that interfere with H1N1 influenza A viral replication. ChemMedChem 2012, 7, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Gong, L.I.; Baltimore, D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010, 328, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Bloom, J.D.; Nayak, J.S.; Baltimore, D. A computational-experimental approach identifies mutations that enhance surface expression of an oseltamivir-resistant influenza neuraminidase. PLoS ONE 2011, 6, e22201. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.A.; Crowe, J.E., Jr.; Bloom, J.D. Influenza viruses with receptor-binding N1 neuraminidases occur sporadically in several lineages and show no attenuation in cell culture or mice. J. Virol. 2015, 89, 3737–3745. [Google Scholar] [CrossRef] [PubMed]
- Victor, S.T.; Watanabe, S.; Katsura, H.; Ozawa, M.; Kawaoka, Y. A replication-incompetent PB2-knockout influenza A virus vaccine vector. J. Virol. 2012, 86, 4123–4128. [Google Scholar] [CrossRef]
- Uraki, R.; Kiso, M.; Iwatsuki-Horimoto, K.; Fukuyama, S.; Takashita, E.; Ozawa, M.; Kawaoka, Y. A novel bivalent vaccine based on a PB2-knockout influenza virus protects mice from pandemic H1N1 and highly pathogenic H5N1 virus challenges. J. Virol. 2013, 87, 7874–7881. [Google Scholar] [CrossRef]
- Kobayashi, H.; Iwatsuki-Horimoto, K.; Kiso, M.; Uraki, R.; Ichiko, Y.; Takimoto, T.; Kawaoka, Y. A replication-incompetent influenza virus bearing the HN glycoprotein of human parainfluenza virus as a bivalent vaccine. Vaccine 2013, 31, 6239–6246. [Google Scholar] [CrossRef]
- Fonseca, W.; Ozawa, M.; Hatta, M.; Orozco, E.; Martinez, M.B.; Kawaoka, Y. A recombinant influenza virus vaccine expressing the F protein of respiratory syncytial virus. Arch. Virol. 2014, 159, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Guo, H.; Albrecht, R.A.; Garcia-Sastre, A.; Topham, D.J.; Martinez-Sobrido, L. Protection against lethal influenza with a viral mimic. J. Virol. 2013, 87, 8591–8605. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Baker, S.F.; Martinez-Sobrido, L.; Topham, D.J. Induction of CD8 T cell heterologous protection by a single dose of single-cycle infectious influenza virus. J. Virol. 2014, 88, 12006–12016. [Google Scholar] [CrossRef] [PubMed]
- Powell, T.J.; Silk, J.D.; Sharps, J.; Fodor, E.; Townsend, A.R. Pseudotyped influenza A virus as a vaccine for the induction of heterotypic immunity. J. Virol. 2012, 86, 13397–13406. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Iwatsuki-Horimoto, K.; Fukuyama, S.; Watanabe, S.; Sakabe, S.; Hatta, Y.; Murakami, S.; Shimojima, M.; Horimoto, T.; Kawaoka, Y. A replication-incompetent virus possessing an uncleavable hemagglutinin as an influenza vaccine. Vaccine 2012, 30, 6027–6033. [Google Scholar] [CrossRef] [PubMed]
- Katsura, H.; Piao, Z.; Iwatsuki-Horimoto, K.; Akeda, Y.; Watanabe, S.; Horimoto, T.; Oishi, K.; Kawaoka, Y. A bivalent vaccine based on a replication-incompetent influenza virus protects against Streptococcus pneumoniae and influenza virus infection. J. Virol. 2014, 88, 13410–13417. [Google Scholar] [CrossRef]
- Shinya, K.; Fujii, Y.; Ito, H.; Ito, T.; Kawaoka, Y. Characterization of a neuraminidase-deficient influenza a virus as a potential gene delivery vector and a live vaccine. J. Virol. 2004, 78, 3083–3088. [Google Scholar] [CrossRef]
- Masic, A.; Pyo, H.M.; Babiuk, S.; Zhou, Y. An eight-segment swine influenza virus harboring H1 and H3 hemagglutinins is attenuated and protective against H1N1 and H3N2 subtypes in pigs. J. Virol. 2013, 87, 10114–10125. [Google Scholar] [CrossRef]
- Pyo, H.M.; Zhou, Y. Protective efficacy of intranasally administered bivalent live influenza vaccine and immunological mechanisms underlying the protection. Vaccine 2014, 32, 3835–3842. [Google Scholar] [CrossRef]
- Hatta, Y.; Boltz, D.; Sarawar, S.; Kawaoka, Y.; Neumann, G.; Bilsel, P. M2SR, a novel live influenza vaccine, protects mice and ferrets against highly pathogenic avian influenza. Vaccine 2017, 35, 4177–4183. [Google Scholar] [CrossRef]
- Sarawar, S.; Hatta, Y.; Watanabe, S.; Dias, P.; Neumann, G.; Kawaoka, Y.; Bilsel, P. M2SR, a novel live single replication influenza virus vaccine, provides effective heterosubtypic protection in mice. Vaccine 2016, 34, 5090–5098. [Google Scholar] [CrossRef]
- Rimmelzwaan, G.F.; Verburgh, R.J.; Nieuwkoop, N.J.; Bestebroer, T.M.; Fouchier, R.A.; Osterhaus, A.D. Use of GFP-expressing influenza viruses for the detection of influenza virus A/H5N1 neutralizing antibodies. Vaccine 2011, 29, 3424–3430. [Google Scholar] [CrossRef]
- Baker, S.F.; Nogales, A.; Santiago, F.W.; Topham, D.J.; Martinez-Sobrido, L. Competitive detection of influenza neutralizing antibodies using a novel bivalent fluorescence-based microneutralization assay (BiFMA). Vaccine 2015, 33, 3562–3570. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Moorthy, N.S.; Poongavanam, V.; Pratheepa, V. Viral M2 ion channel protein: A promising target for anti-influenza drug discovery. Mini Rev. Med. Chem. 2014, 14, 819–830. [Google Scholar]
- Watanabe, S.; Watanabe, T.; Kawaoka, Y. Influenza A virus lacking M2 protein as a live attenuated vaccine. J. Virol. 2009, 83, 5947–5950. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Watanabe, S.; Kim, J.H.; Hatta, M.; Kawaoka, Y. Novel approach to the development of effective H5N1 influenza A virus vaccines: Use of M2 cytoplasmic tail mutants. J. Virol. 2008, 82, 2486–2492. [Google Scholar] [CrossRef] [PubMed]
- Hatta, Y.; Hatta, M.; Bilsel, P.; Neumann, G.; Kawaoka, Y. An M2 cytoplasmic tail mutant as a live attenuated influenza vaccine against pandemic (H1N1) 2009 influenza virus. Vaccine 2011, 29, 2308–2312. [Google Scholar] [CrossRef]
- Si, L.; Xu, H.; Zhou, X.; Zhang, Z.; Tian, Z.; Wang, Y.; Wu, Y.; Zhang, B.; Niu, Z.; Zhang, C.; et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science 2016, 354, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Normanly, J.; Kleina, L.G.; Masson, J.M.; Abelson, J.; Miller, J.H. Construction of Escherichia coli amber suppressor tRNA genes. III. Determination of tRNA specificity. J. Mol. Biol. 1990, 213, 719–726. [Google Scholar] [CrossRef]
- Erbelding, E.J.; Post, D.J.; Stemmy, E.J.; Roberts, P.C.; Augustine, A.D.; Ferguson, S.; Paules, C.I.; Graham, B.S.; Fauci, A.S. A Universal Influenza Vaccine: The Strategic Plan for the National Institute of Allergy and Infectious Diseases. J. Infect. Dis. 2018, 218, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Sautto, G.A.; Kirchenbaum, G.A.; Ross, T.M. Towards a universal influenza vaccine: Different approaches for one goal. Virol. J. 2018, 15, 17. [Google Scholar] [CrossRef] [PubMed]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Lobo, P.; Nogales, A.; Rodríguez, L.; Martínez-Sobrido, L. Novel Approaches for The Development of Live Attenuated Influenza Vaccines. Viruses 2019, 11, 190. https://doi.org/10.3390/v11020190
Blanco-Lobo P, Nogales A, Rodríguez L, Martínez-Sobrido L. Novel Approaches for The Development of Live Attenuated Influenza Vaccines. Viruses. 2019; 11(2):190. https://doi.org/10.3390/v11020190
Chicago/Turabian StyleBlanco-Lobo, Pilar, Aitor Nogales, Laura Rodríguez, and Luis Martínez-Sobrido. 2019. "Novel Approaches for The Development of Live Attenuated Influenza Vaccines" Viruses 11, no. 2: 190. https://doi.org/10.3390/v11020190
APA StyleBlanco-Lobo, P., Nogales, A., Rodríguez, L., & Martínez-Sobrido, L. (2019). Novel Approaches for The Development of Live Attenuated Influenza Vaccines. Viruses, 11(2), 190. https://doi.org/10.3390/v11020190