Duck Plague Virus Promotes DEF Cell Apoptosis by Activating Caspases, Increasing Intracellular ROS Levels and Inducing Cell Cycle S-Phase Arrest

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Cell Viability
2.3. Detection of DPV
2.4. DAPI Staining of Cell Nuclei
2.5. Isolation of RNA and Analysis of the mRNA Expression of Apoptosis-Related Genes by qRT-PCR
2.6. Flow Cytometric Analysis of Apoptosis
2.7. FCM Cell Cycle Analysis
2.8. Determination of Caspase-3, Caspase-7, Caspase-8, and Caspase-9 Activities
2.9. Determination of MMP
2.10. Determination of Intracellular ROS Levels
2.11. Statistical Analysis
3. Results
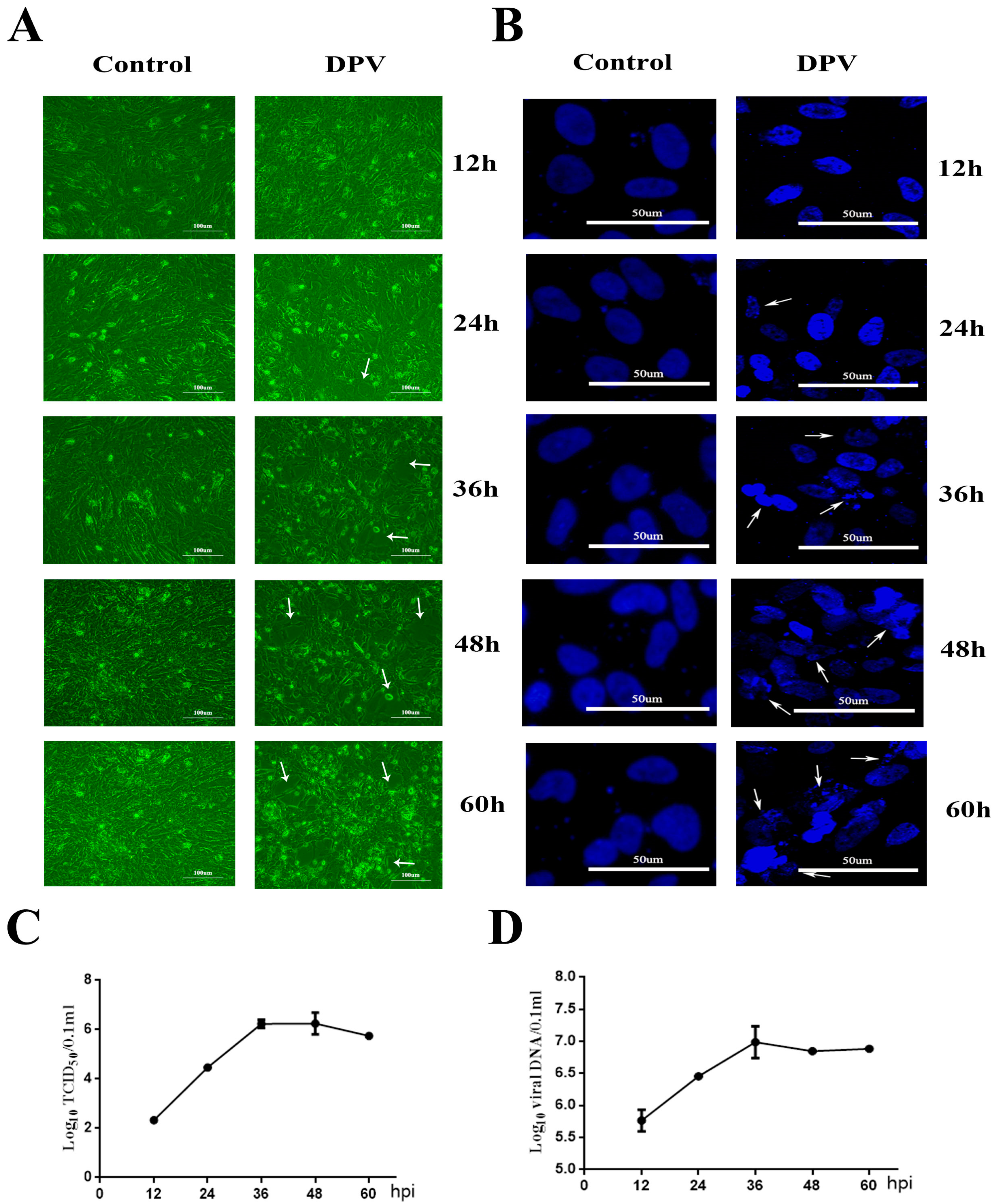
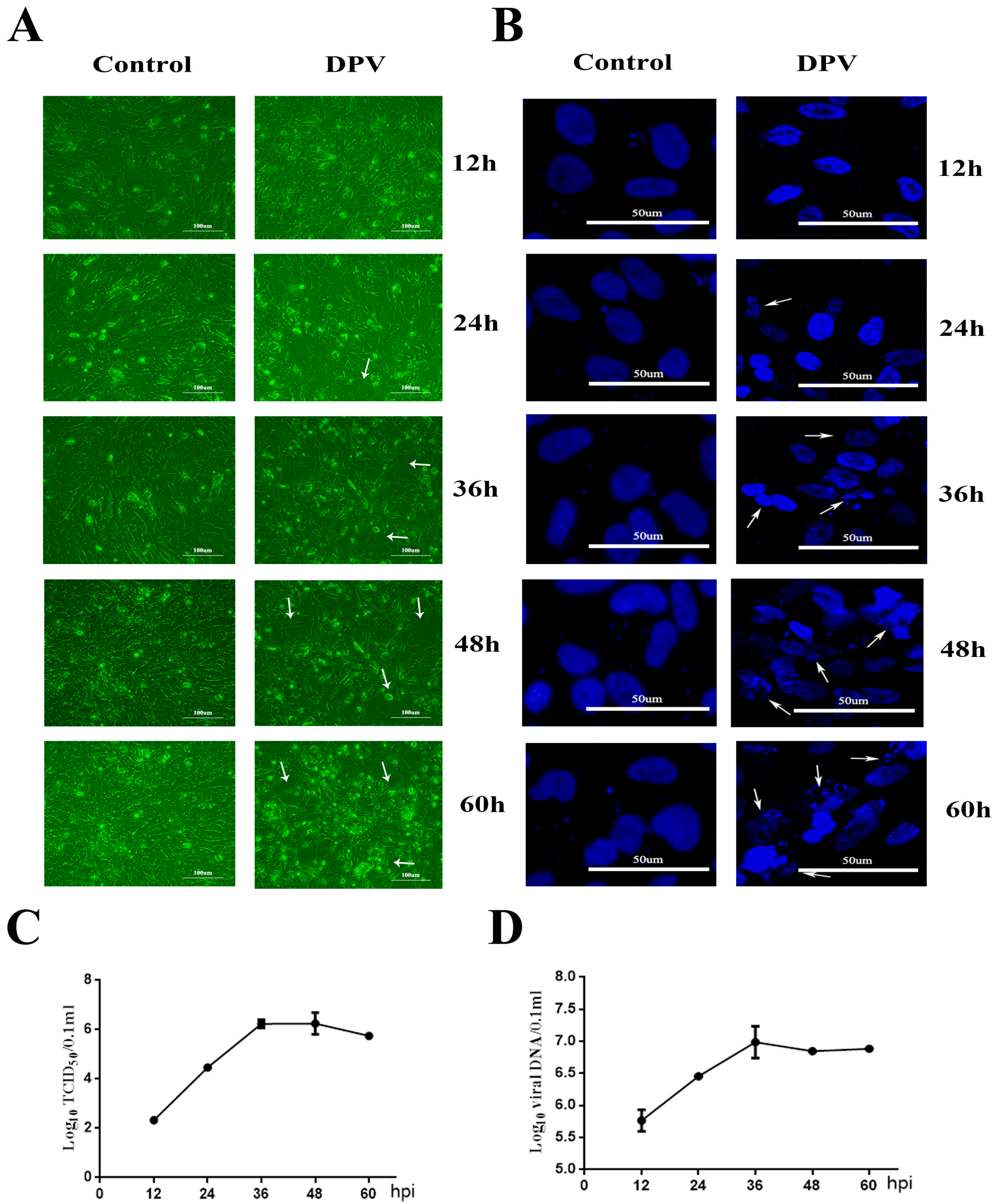
3.1. Cytopathic Effects (CPEs) Induced by DPV in DEFs
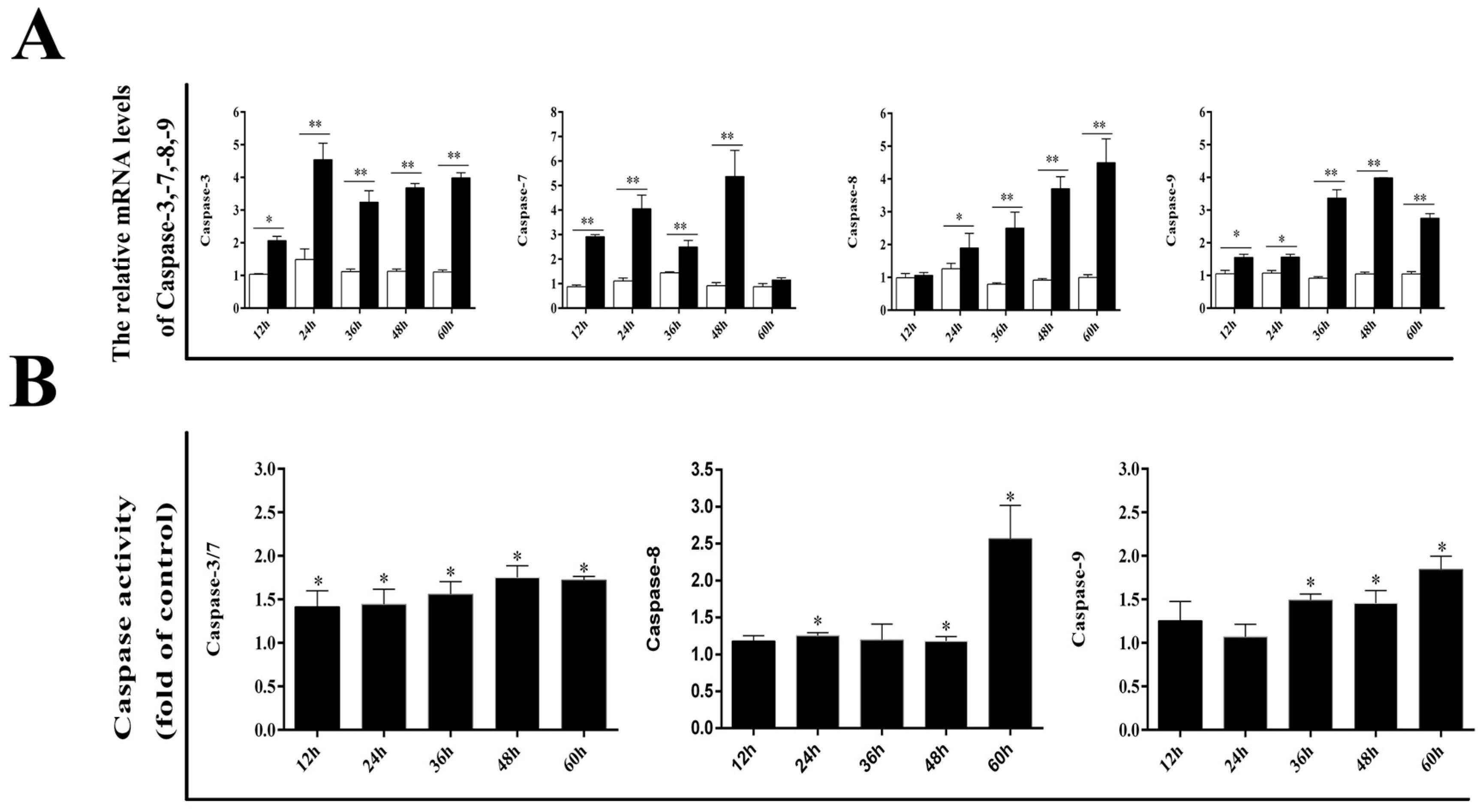
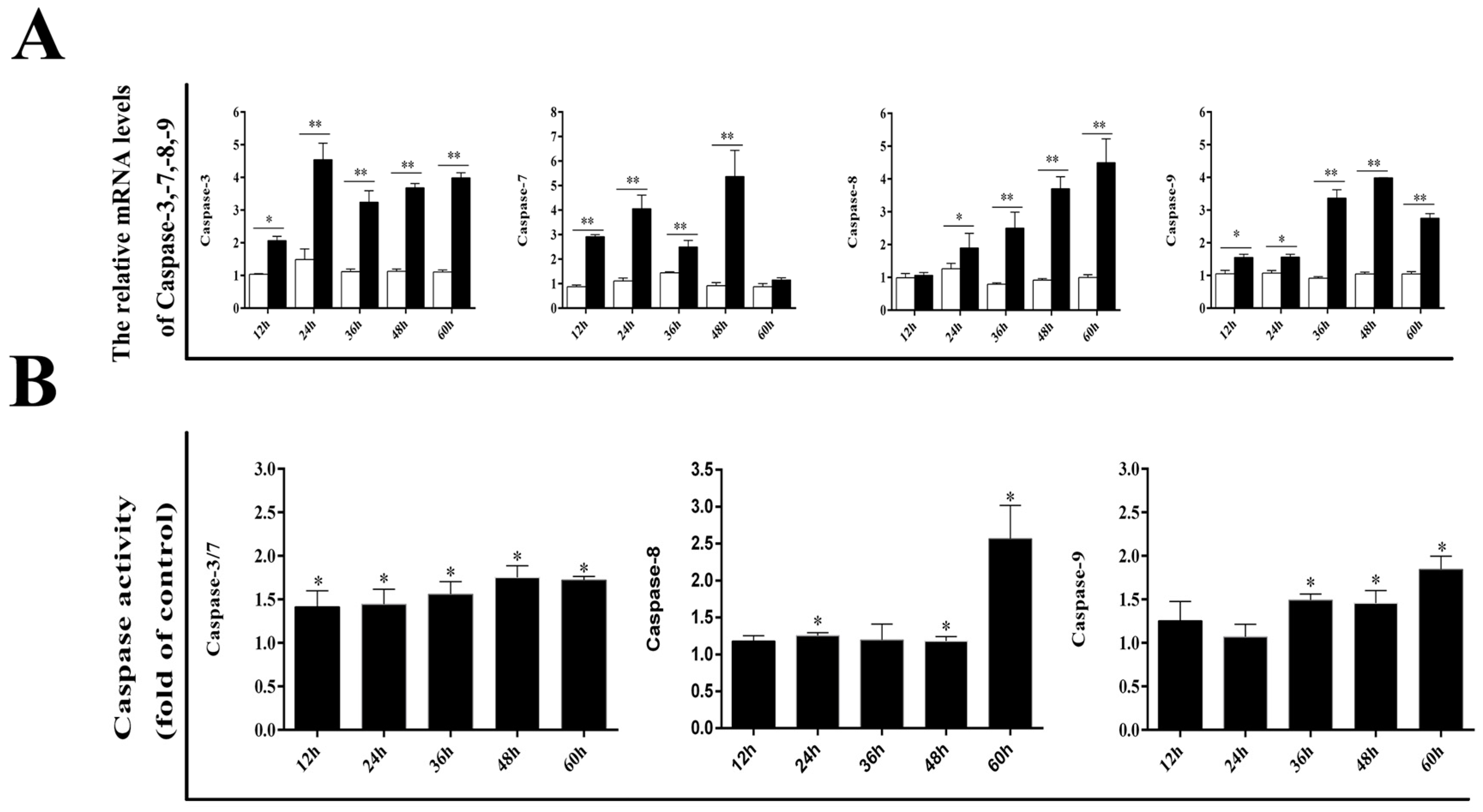
3.2. Effect of DPV Infection on Caspases
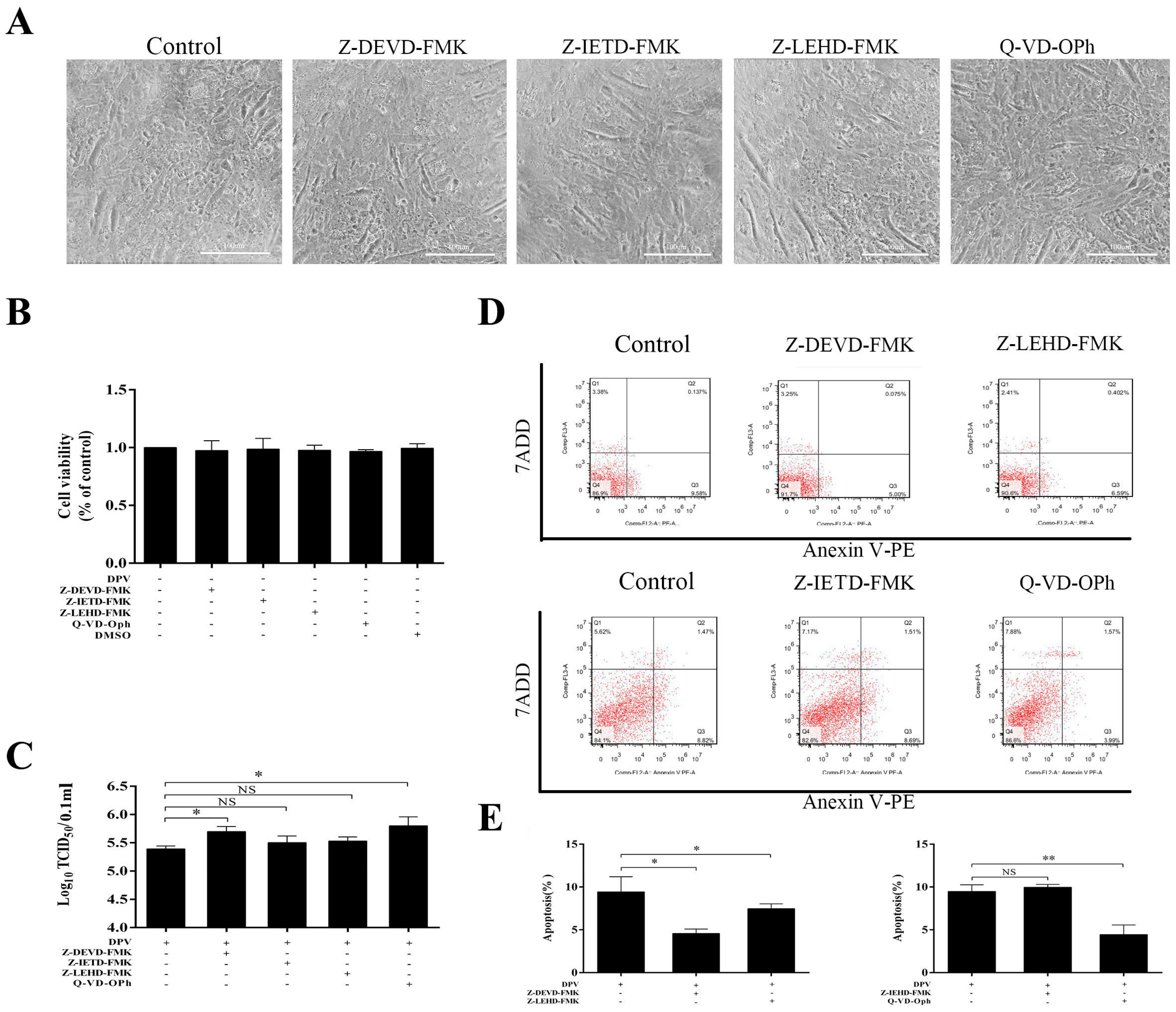
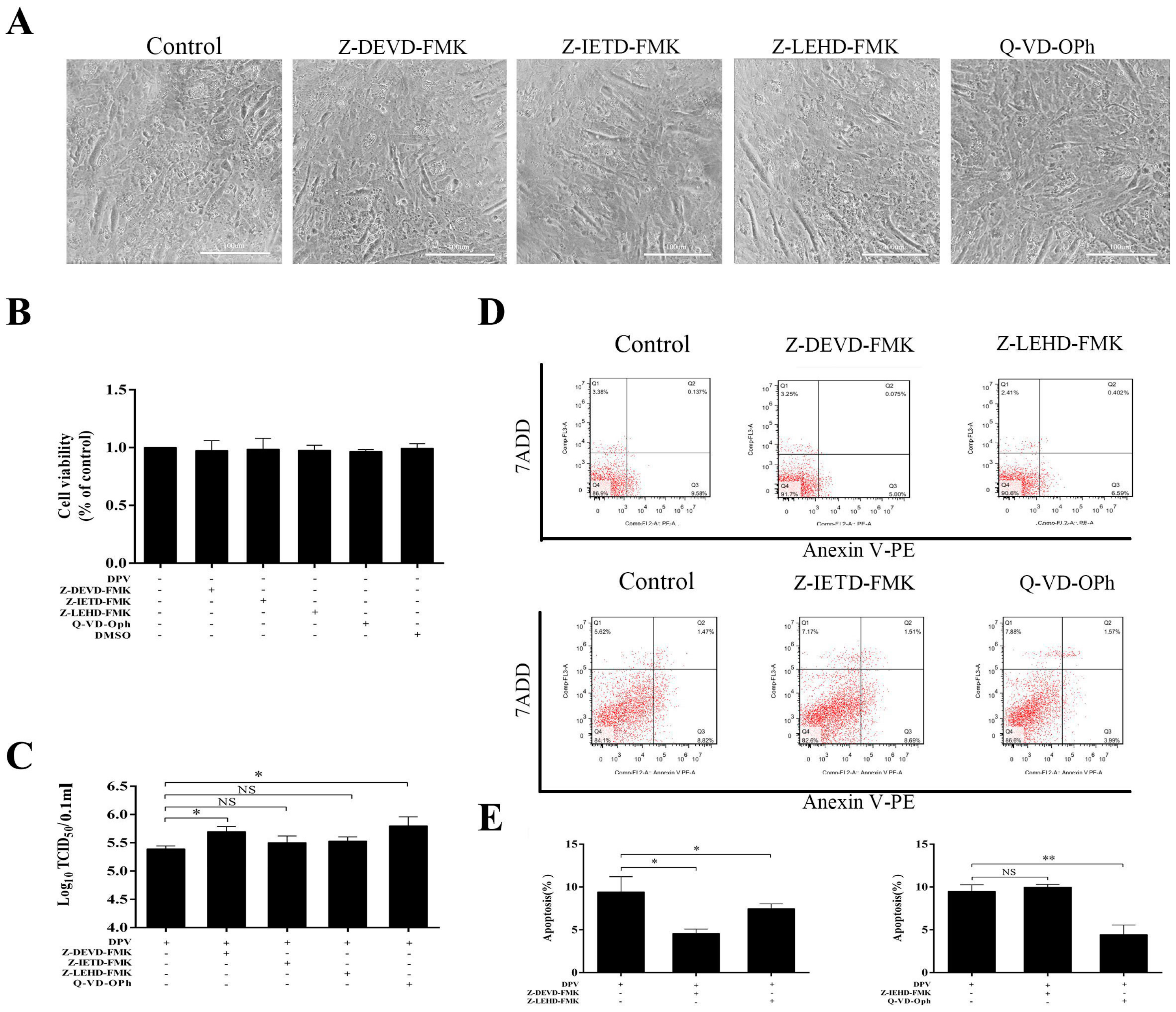
3.3. Caspase Inhibitors Inhibit DPV-Induced Apoptosis and Promote Viral Replication
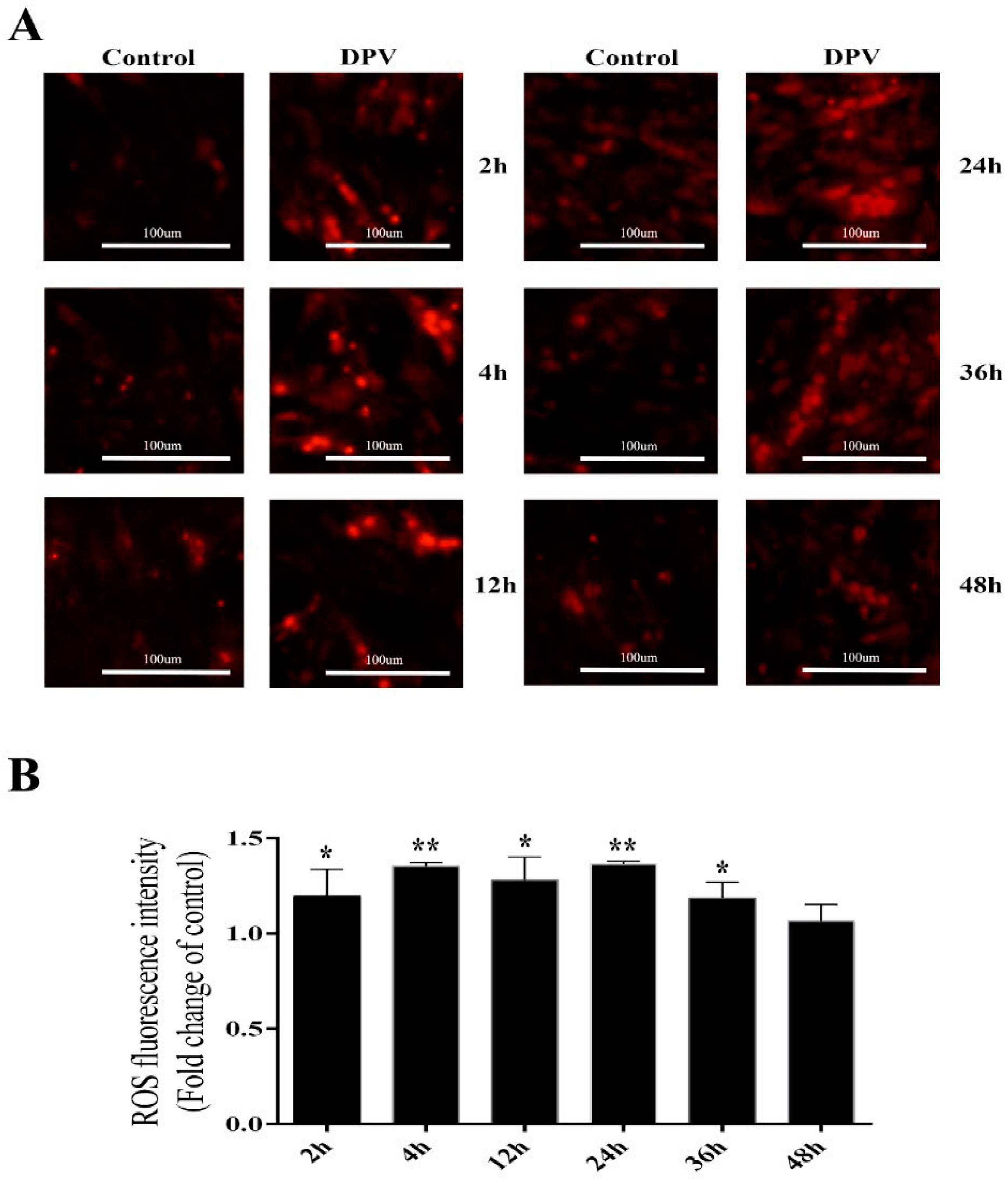
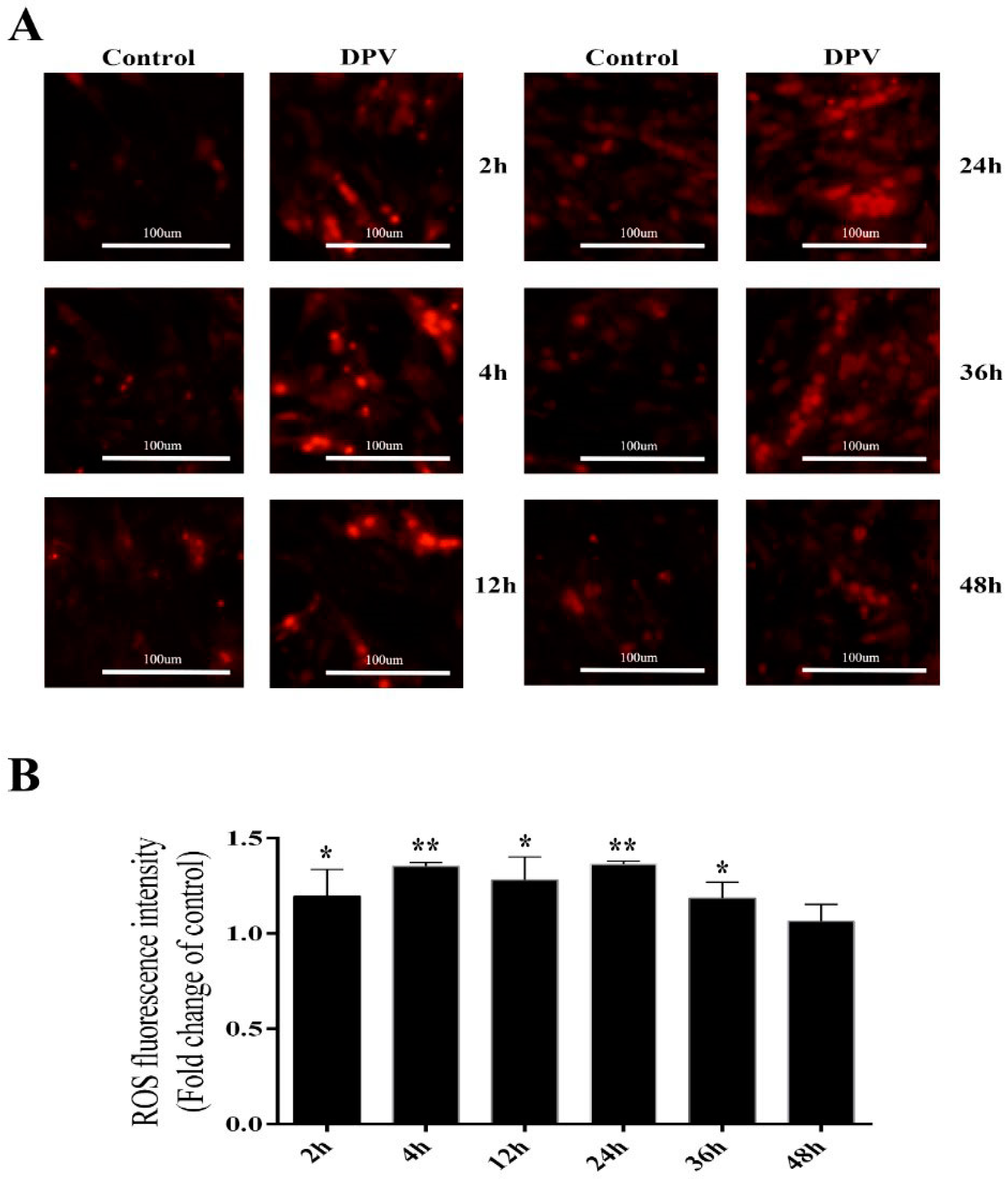
3.4. Effect of DPV Infection on ROS
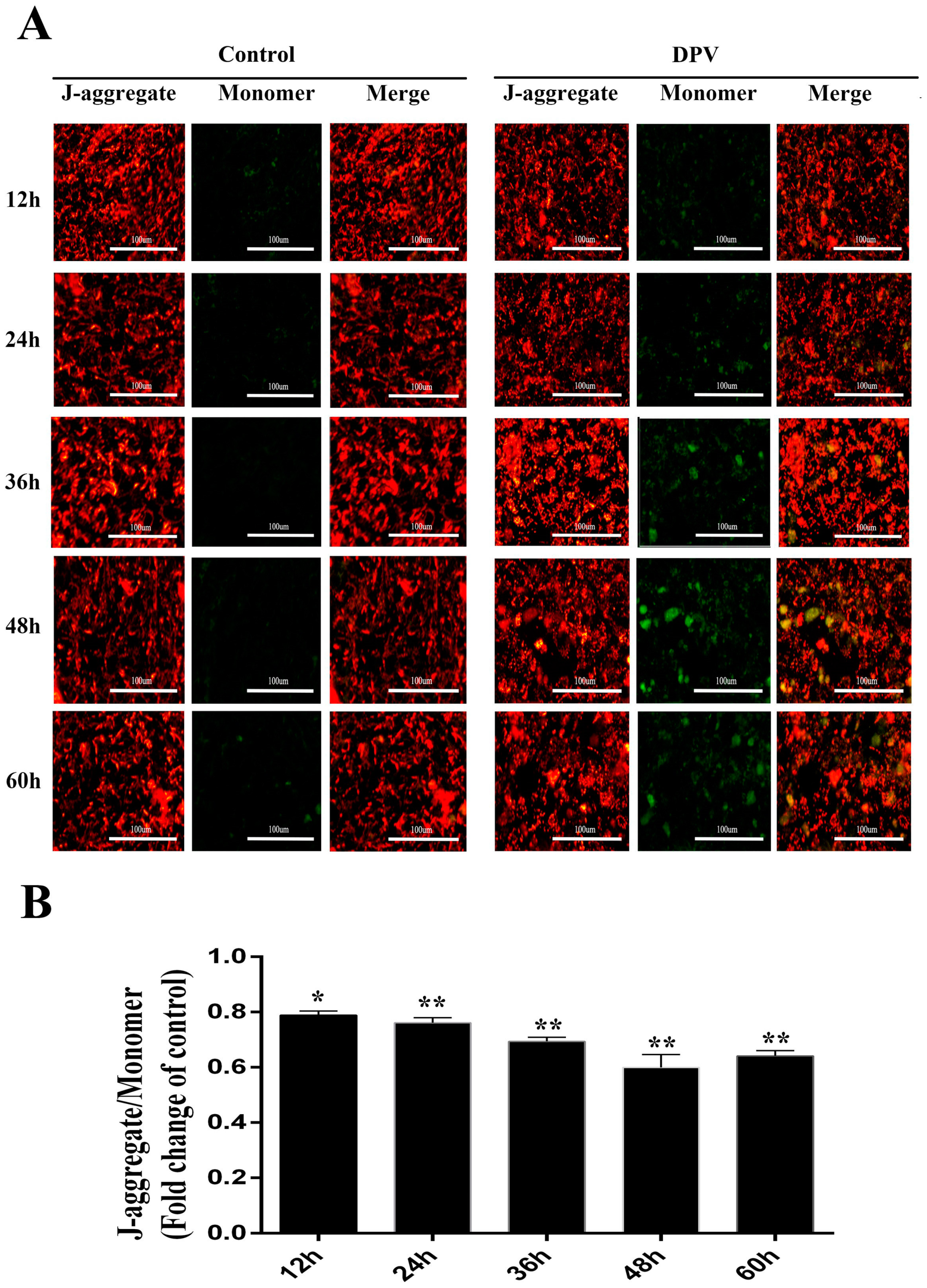
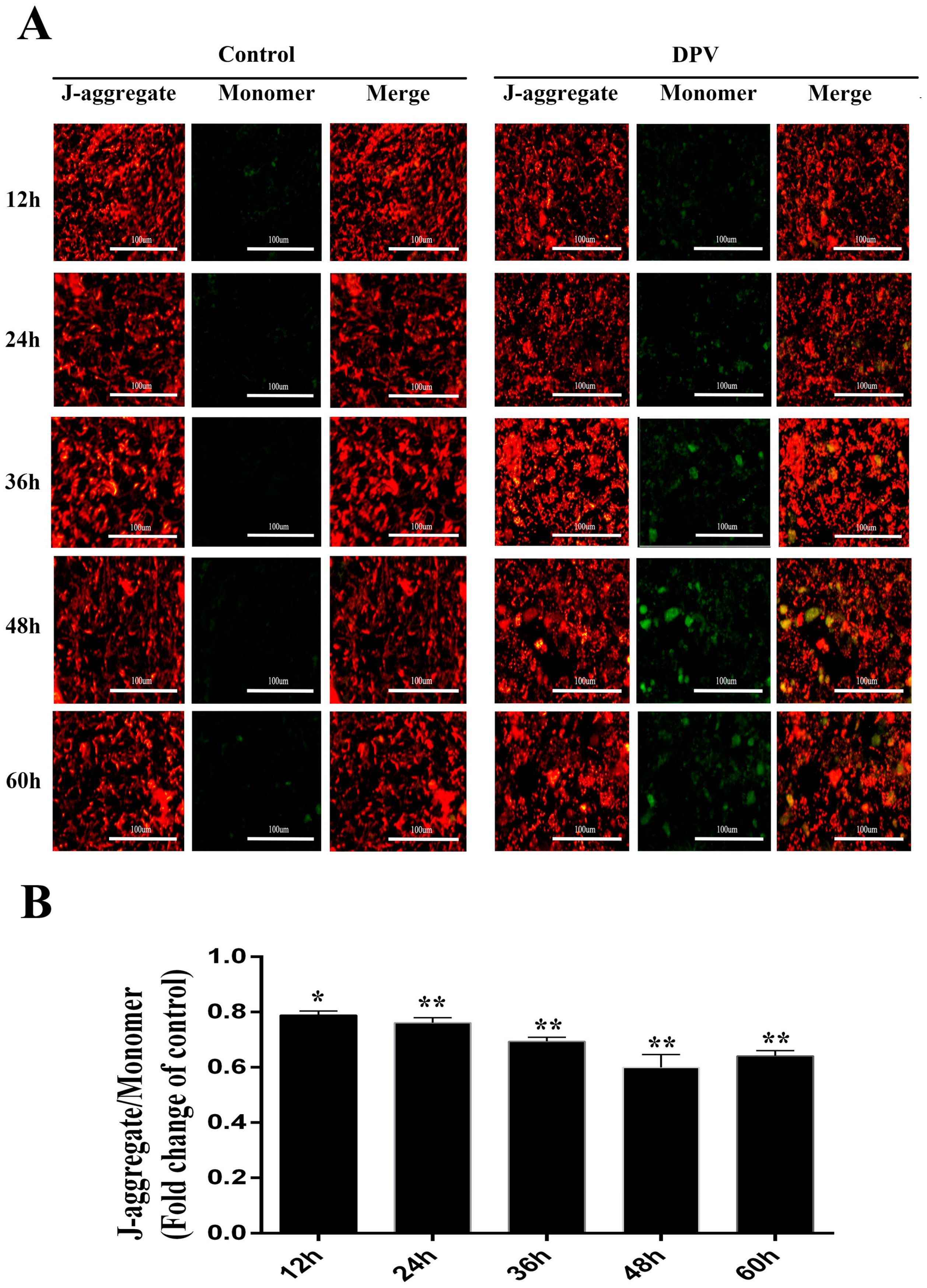
3.5. Effect of DPV Infection on MMP
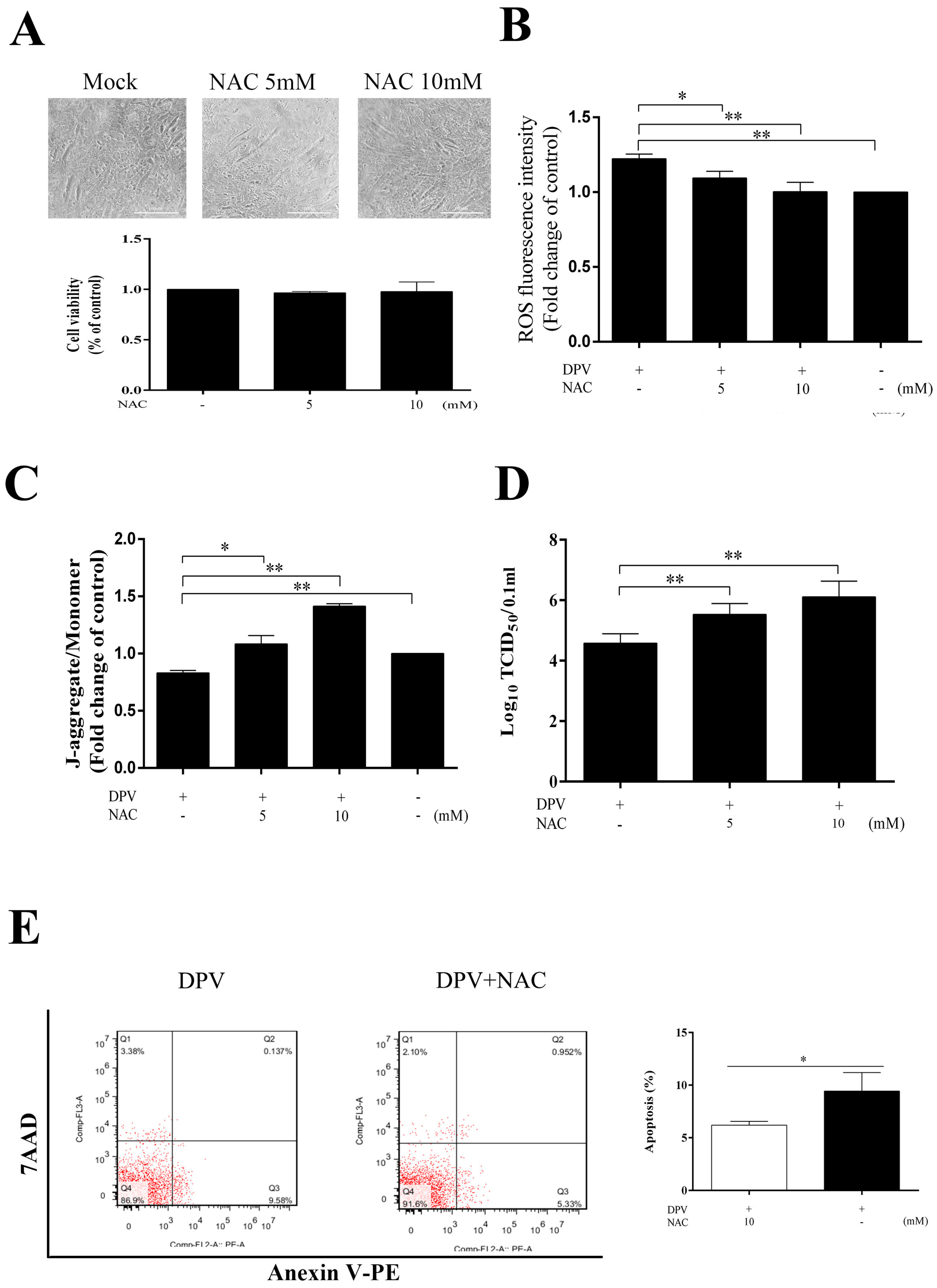
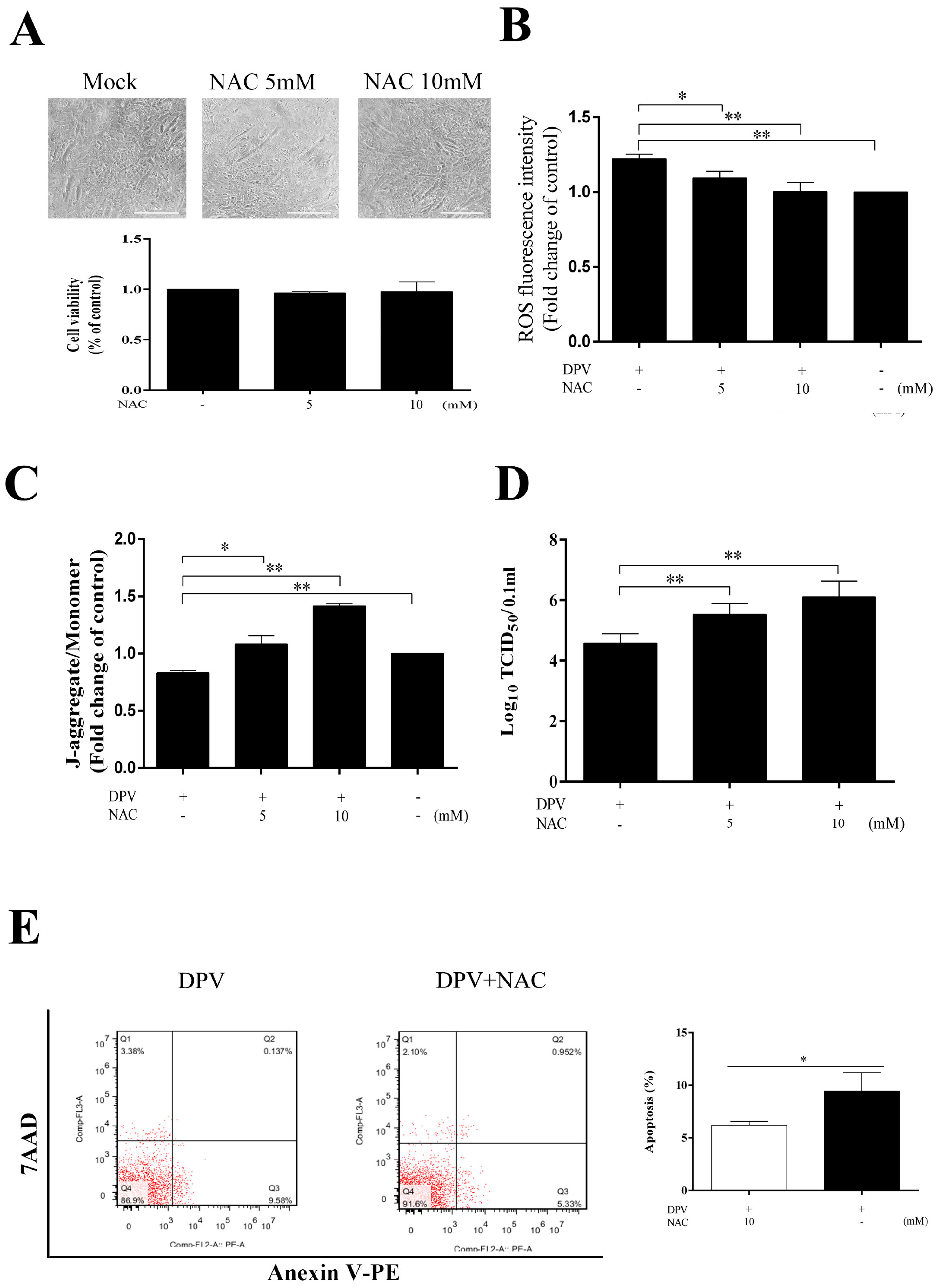
3.6. NAC Scavenges Intracellular ROS, Increases the MMP, Inhibits Apoptosis and Promotes Viral Replication
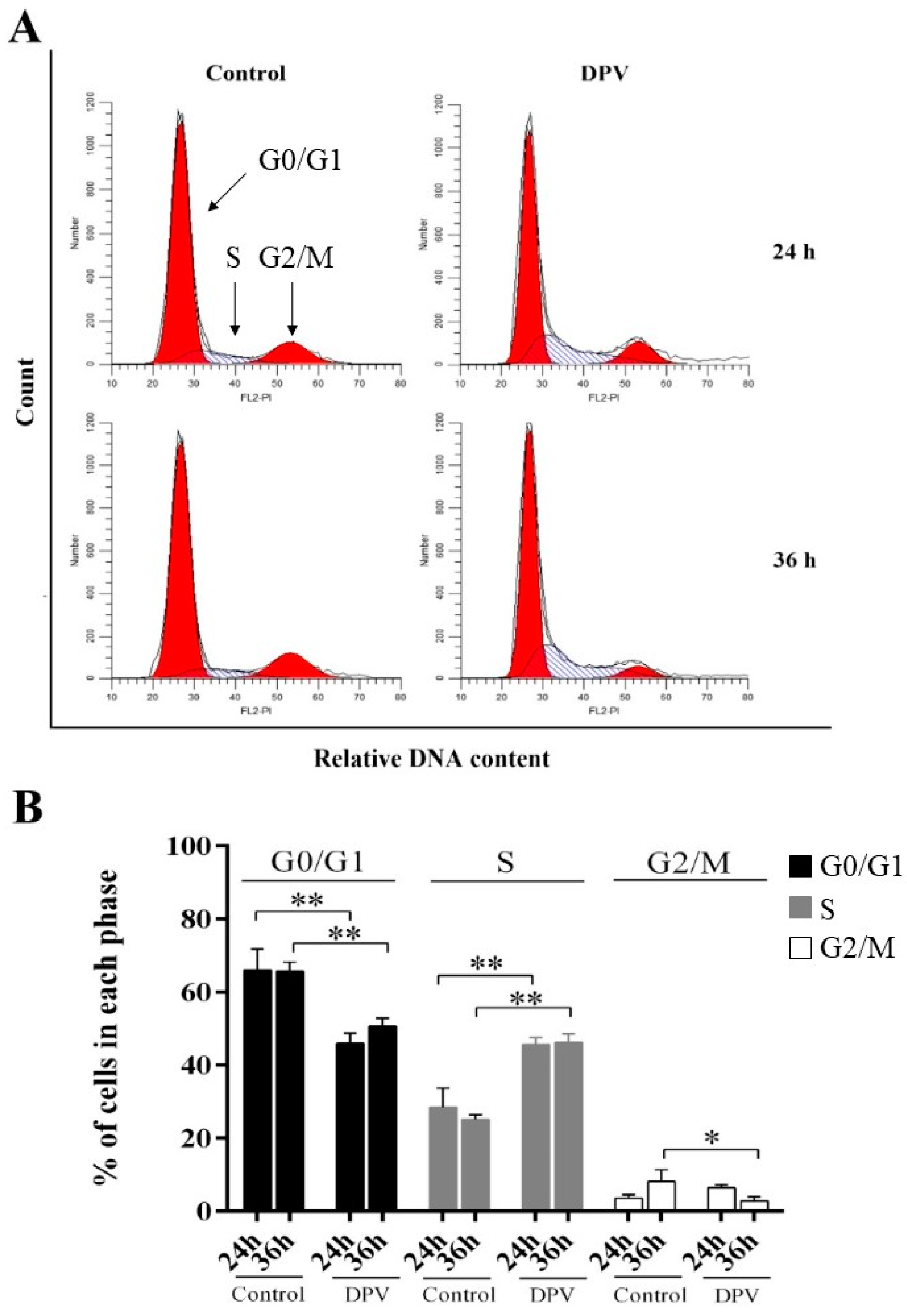
3.7. DPV Induces DEF S-Phase Cell Cycle Arrest
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Anchun, C.; Mingshu, W.; Ming, W.; Weiguang, Z.; Yufei, G.; Renyong, J.; Chao, X.; Guiping, Y.; Yicheng, L. Construction of duck enteritis virus gene libraries and discovery, cloning and identification of viral nucleocapsid protein gene. Chin. High Technol. Lett. 2006, 16, 948–953. [Google Scholar]
- Yuan, G.P.; Cheng, A.C.; Wang, M.S.; Liu, F.; Han, X.Y.; Liao, Y.H.; Xu, C. Electron microscopic studies of the morphogenesis of duck enteritis virus. Avian Dis. 2005, 49, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, A.; Wang, M.; Zhu, D.; Jia, R.; Chen, S.; Zhou, Y.; Chen, X. Comparative genomic analysis of duck enteritis virus strains. J. Virol. 2012, 86, 13841–13842. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cheng, A.; Wang, M.; Yang, Q.; Zhu, D.; Jia, R.; Chen, S.; Zhou, Y.; Wang, X.; Chen, X. Complete genomic sequence of chinese virulent duck enteritis virus. J. Virol. 2012, 86, 5965. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, X.; Cheng, A.; Wang, M.; Zhu, D.; Jia, R.; Luo, Q.; Chen, X. Intestinal Mucosal Immune Response against Virulent Duck Enteritis Virus Infection in Ducklings. Res. Vet. Sci. 2008, 87, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Cheng, A.; Wang, M.; Zhou, Y. Purification of anatid herpesvirus 1 particles by tangential-flow ultrafiltration and sucrose gradient ultracentrifugation. J. Virol. Methods 2009, 161, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Yang, X.Y.; Cheng, A.C.; Wang, M.; Zhu, D.; Jia, R. The pathogenesis of duck virus enteritis in experimentally infected ducks: A quantitative time-course study using TaqMan polymerase chain reaction. Poult. Sci. 2008, 87, 1768–1772. [Google Scholar]
- Qi, X.; Yang, X.; Cheng, A.; Wang, M.; Zhu, D.; Jia, R. Quantitative analysis of virulent duck enteritis virus loads in experimentally infected ducklings. Avian Dis. 2008, 52, 338–344. [Google Scholar] [PubMed]
- Guiping, Y.; Anchun, C.; Mingshu, W.; Yi, Z.; Fei, L.; Yufei, G.; Xiaoying, H.; Yonghong, L.; Chao, X.; Weiguang, Z.; et al. Ultrastructural pathological observation on ducks infected expeimentally with duck enteritis virus. Chin. J. Vet. Sci. 2005, 25, 459–462. [Google Scholar]
- Anchun, C. Duck Plague; China Agriculture Press: Beijing, China, 2015. [Google Scholar]
- Yang, X.; Qi, X.; Cheng, A.; Wang, M.; Zhu, D.; Jia, R.; Chen, X. Intestinal mucosal immune response in ducklings following oral immunisation with an attenuated duck enteritis virus vaccine. Vet. J. 2010, 185, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Jia, R.; Huang, J.; Shu, B.; Zhu, D.; Liu, Q.; Gao, X.; Meng, L.; Yin, Z.; Wang, M. Attenuated salmonella typhimurium delivering DNA vaccine encoding duck enteritis virus UL24 induced systemic and mucosal immune responses and conferred good protection against challenge. Vet. Res. 2012, 43, 56. [Google Scholar]
- Sun, K.; Xin, L.; Jiang, J.; Cheng, A.; Wang, M.; Zhu, D.; Jia, R.; Chen, S.; Yi, Z.; Chen, X. Distribution characteristics of DNA vaccine encoded with glycoprotein c from anatid herpesvirus 1 with chitosan and liposome as deliver carrier in ducks. Virol. J. 2013, 10, 89. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Yang, X.; Cheng, A.; Wang, M.; Guo, Y.; Jia, R. Replication kinetics of duck virus enteritis vaccine virus in ducklings immunized by the mucosal or systemic route using real-time quantitative PCR. Res. Vet. Sci. 2009, 86, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.F.; Cheng, A.C.; Wang, M.S.; Yang, X.Y.; Jia, R.Y.; Chen, X.Y. Induction of IGA, IGM and IGG antibodies in ducklings immunized with attenuated duck plague virus vaccine strain. Sci. Agric. Sin. 2008, 41, 3305–3310. [Google Scholar]
- Anchun, C.; Mingshu, W.; Fei, L.; Yong, S.; Guiping, Y.; Xiaoying, H.; Chao, X.; Yonghong, L.; Ming, W.; Weiguang, Z. Distribution and excretion of duck plague virus attenuated cha strain in vaccinated ducklings. Chin. J. Vet. Sci. 2005, 25, 231–233. [Google Scholar]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Cheng, A.C.; Wang, M.S.; Jia, R.Y.; Sun, K.F.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; Liu, M.F.; et al. The suppression of apoptosis by alpha-herpesvirus. Cell Death Dis. 2017, 8, e2749. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.H.; Viacava Follis, A.; Kriwacki, R.W.; Moldoveanu, T. Discoveries and controversies in BCL-2 protein-mediated apoptosis. FEBS J. 2016, 283, 2690–2700. [Google Scholar] [CrossRef] [PubMed]
- Shore, G.C.; Papa, F.R.; Oakes, S.A. Signaling cell death from the endoplasmic reticulum stress response. Curr. Opin. Cell Biol. 2011, 23, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Verma, G.; Datta, M. The critical role of JNK in the ER-mitochondrial crosstalk during apoptotic cell death. J. Cell. Physiol. 2012, 227, 1791–1795. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Malik, A.; Holohan, C.; Longley, D.B. Ded or alive: Assembly and regulation of the death effector domain complexes. Cell Death Dis. 2015, 6, e1866. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Shen, C.; Cheng, A.; Wang, M.; Zhang, N.; Chen, S.; Zhou, Y. Anatid herpesvirus 1 ch virulent strain induces syncytium and apoptosis in duck embryo fibroblast cultures. Vet. Microbiol. 2009, 138, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Guiping, Y.; Anchun, C.; Mingshu, W.; Xiaoying, H.; Yi, Z.; Fei, L. Preliminary study on duck enteritis virus-induced lymphocyte apoptosis in vivo. Avian Dis. 2007, 51, 546–549. [Google Scholar] [CrossRef]
- Zhao, L.; Cheng, A.; Wang, M.; Yuan, G.; Cai, M. Characterization of codon usage bias in the dutpase gene of duck enteritis virus. Prog. Nat. Sci. Mater. Int. 2008, 18, 1069–1076. [Google Scholar] [CrossRef]
- Xin, H.Y.; Cheng, A.C.; Wang, M.S.; Jia, R.Y.; Shen, C.J.; Chang, H. Identification and characterization of a duck enteritis virus US3-like gene. Avian Dis. 2009, 53, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Yufei, G.; Anchun, C.; Mingshu, W.; Renyong, J.; Ming, W.; Weiguang, Z.; Xiaoyue, C. Establishment and application of quantitative real time pcr assay for detecting duck viral enteritis virus. Chin. J. Vet. Sci. 2006, 36, 444–448. [Google Scholar]
- You, Y.; Liu, T.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; Liu, M.; et al. Duck plague virus glycoprotein j is functional but slightly impaired in viral replication and cell-to-cell spread. Sci. Rep. 2018, 8, 4069. [Google Scholar] [CrossRef] [PubMed]
- Tsalenchuck, Y.; Steiner, I.; Panet, A. Innate defense mechanisms against HSV-1 infection in the target tissues, skin and brain. J. Neurovirol. 2016, 22, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Geiser, V.; Rose, S.; Jones, C. Bovine herpesvirus type 1 induces cell death by a cell-type-dependent fashion. Microb. Pathog. 2008, 44, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.J.; Lin, P.Y.; Liao, M.H.; Liu, H.J.; Lee, J.W.; Chiu, S.J.; Hsu, H.Y.; Shih, W.L. TNF-α mediates pseudorabies virus-induced apoptosis via the activation of p38 MAPK and JNK/SAPK signaling. Virology 2008, 381, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Longo, M.; Fiorito, F.; Marfe, G.; Montagnaro, S.; Pisanelli, G.; De Martino, L.; Iovane, G.; Pagnini, U. Analysis of apoptosis induced by caprine herpesvirus 1 in vitro. Virus Res. 2009, 145, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Gillis, P.A.; Okagaki, L.H.; Rice, S.A. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J. Virol. 2009, 83, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, L.R.; Jones, C.J. Activation of caspases and p53 by bovine herpesvirus 1 infection results in programmed cell death and efficient virus release. J. Virol. 1999, 73, 3778–3788. [Google Scholar] [PubMed]
- Xu, X.; Zhang, K.; Huang, Y.; Ding, L.; Chen, G.; Zhang, H.; Tong, D. Bovine herpes virus type 1 induces apoptosis through fas-dependent and mitochondria-controlled manner in madin-darby bovine kidney cells. Virol. J. 2012, 9, 202. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Schachtele, S.J.; Lokensgard, J.R. Reactive oxygen species drive herpes simplex virus (hsv)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflammation. 2011, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Schachtele, S.J.; Hu, S.; Lokensgard, J.R. Modulation of experimental herpes encephalitis-associated neurotoxicity through sulforaphane treatment. PLoS ONE 2012, 7, e36216. [Google Scholar] [CrossRef] [PubMed]
- Kavouras, J.H.; Prandovszky, E.; Valyi-Nagy, K.; Kovacs, S.K.; Tiwari, V.; Kovacs, M.; Shukla, D.; Valyi-Nagy, T. Herpes simplex virus type 1 infection induces oxidative stress and the release of bioactive lipid peroxidation by-products in mouse p19n neural cell cultures. J. Neurovirol. 2007, 13, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dosal, R.; Horan, K.A.; Rahbek, S.H.; Ichijo, H.; Chen, Z.J.; Mieyal, J.J.; Hartmann, R.; Paludan, S.R. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: Role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. 2011, 7, e1002250. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, C.; Zhang, D.; Ma, Y.; Ding, X.; Zhu, G. Bhv-1 induced oxidative stress contributes to mitochondrial dysfunction in mdbk cells. Vet. Res. 2016, 47, 47. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Goshima, F.; Daikoku, T.; Inagaki-Ohara, K.; Takakuwa, H.; Kato, K.; Nishiyama, Y. Mitochondrial distribution and function in herpes simplex virus-infected cells. J. Gen. Virol. 2000, 81, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Choi, S.H.; Kim, J.K.; Kim, Y.; Kim, H.J.; Im, J.S.; Lee, S.Y.; Choi, J.M.; Lee, H.M.; Ahn, J.K. Herpes simplex virus type 1 ICP27 induces apoptotic cell death by increasing intracellular reactive oxygen species. Mol. Biol. 2008, 42, 470–477. [Google Scholar] [CrossRef]
- Hobbs, W.E., 2nd; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar] [PubMed]
- Li, H.; Baskaran, R.; Krisky, D.M.; Bein, K.; Grandi, P.; Cohen, J.B.; Glorioso, J.C. CHK2 is required for hsv-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 2008, 375, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Bringhurst, R.M.; Schaffer, P.A. Cellular stress rather than stage of the cell cycle enhances the replication and plating efficiencies of herpes simplex virus type 1 ICP0- viruses. J. Virol. 2006, 80, 4528–4537. [Google Scholar] [CrossRef] [PubMed]
- Pagnini, U.; Montagnaro, S.; Sanfelice di Monteforte, E.; Pacelli, F.; De Martino, L.; Roperto, S.; Florio, S.; Iovane, G. Caprine herpesvirus-1 (CAPHV-1) induces apoptosis in goat peripheral blood mononuclear cells. Vet. Immunol. Immunopathol. 2005, 103, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Trapp-Fragnet, L.; Bencherit, D.; Chabanne-Vautherot, D.; Le Vern, Y.; Remy, S.; Boutet-Robinet, E.; Mirey, G.; Vautherot, J.F.; Denesvre, C. Cell cycle modulation by marek’s disease virus: The tegument protein VP22 triggers s-phase arrest and DNA damage in proliferating cells. PLoS ONE 2014, 9, e100004. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer Sequences | Gene Accession Number | |
|---|---|---|---|
| Caspase-3 | For | 5′ TGGTGTTGAGGCAGACAGTGGA 3′ | XM_005030494 |
| Rev | 5′ CATTCCGCCAGGAGTAATAGCC 3′ | ||
| Caspase-8 | For | 5′ GGTGATGCTCGTCAGAAAGGTG 3′ | XM_013094737 |
| Rev | 5′ AGCCATGCCCAAGAGGAAGT 3′ | ||
| Caspase-9 | For | 5′GCTGCTTCAACTTCCTCCGTAA 3′ | XM_013095294 |
| Rev | 5′ CATCTCCACGGACAGACAAAGG 3′ | ||
| β-Actin | For | 5′ CCGGGCATCGCTGACA 3′ | NM_001310421 |
| Rev | 5′ GGATTCATCATACTCCTGCTTTGCT 3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, C.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Zhu, D.; Chen, S.; Liu, M.; Zhao, X.; et al. Duck Plague Virus Promotes DEF Cell Apoptosis by Activating Caspases, Increasing Intracellular ROS Levels and Inducing Cell Cycle S-Phase Arrest. Viruses 2019, 11, 196. https://doi.org/10.3390/v11020196
Zhao C, Wang M, Cheng A, Yang Q, Wu Y, Jia R, Zhu D, Chen S, Liu M, Zhao X, et al. Duck Plague Virus Promotes DEF Cell Apoptosis by Activating Caspases, Increasing Intracellular ROS Levels and Inducing Cell Cycle S-Phase Arrest. Viruses. 2019; 11(2):196. https://doi.org/10.3390/v11020196
Chicago/Turabian StyleZhao, Chuankuo, Mingshu Wang, Anchun Cheng, Qiao Yang, Ying Wu, Renyong Jia, Dekang Zhu, Shun Chen, Mafeng Liu, Xinxin Zhao, and et al. 2019. "Duck Plague Virus Promotes DEF Cell Apoptosis by Activating Caspases, Increasing Intracellular ROS Levels and Inducing Cell Cycle S-Phase Arrest" Viruses 11, no. 2: 196. https://doi.org/10.3390/v11020196
APA StyleZhao, C., Wang, M., Cheng, A., Yang, Q., Wu, Y., Jia, R., Zhu, D., Chen, S., Liu, M., Zhao, X., Zhang, S., Liu, Y., Yu, Y., Zhang, L., Tian, B., Rehman, M. U., Pan, L., & Chen, X. (2019). Duck Plague Virus Promotes DEF Cell Apoptosis by Activating Caspases, Increasing Intracellular ROS Levels and Inducing Cell Cycle S-Phase Arrest. Viruses, 11(2), 196. https://doi.org/10.3390/v11020196