
On the Hunt for Next-Generation Antimicrobial Agents: An Online Symposium Organized Jointly by the French Society for Medicinal Chemistry (Société de Chimie Thérapeutique) and the French Microbiology Society (Société Française de Microbiologie) on 9–10 December 2021

 ,
,  , and
, and 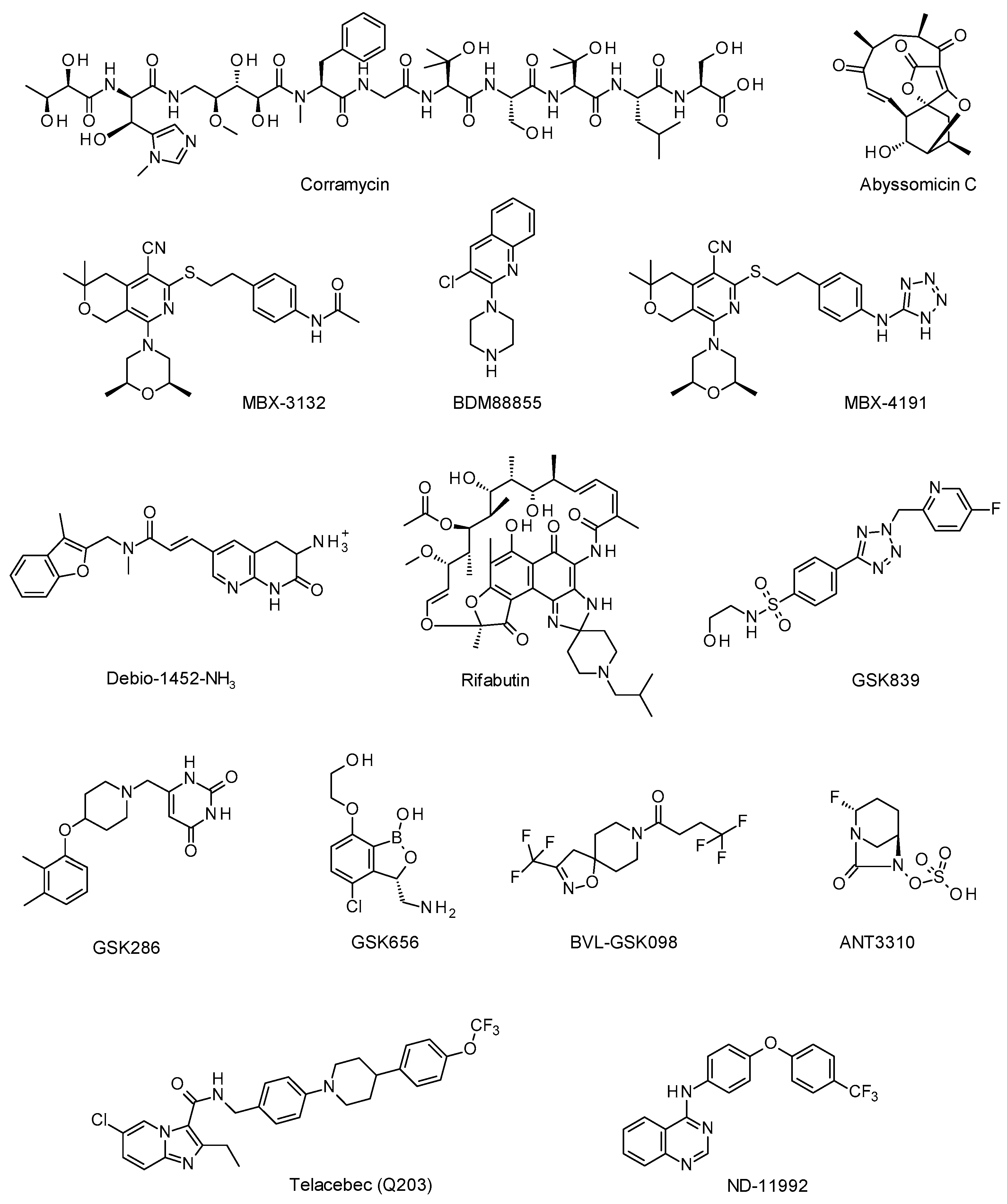{kind=link}
Abstract
:1. Aim and Scope of the Meeting
2. Opening Lecture
3. Keynote Lectures on Thursday, 9 December
3.1. Session 1: The Hunt for Active Natural Compounds
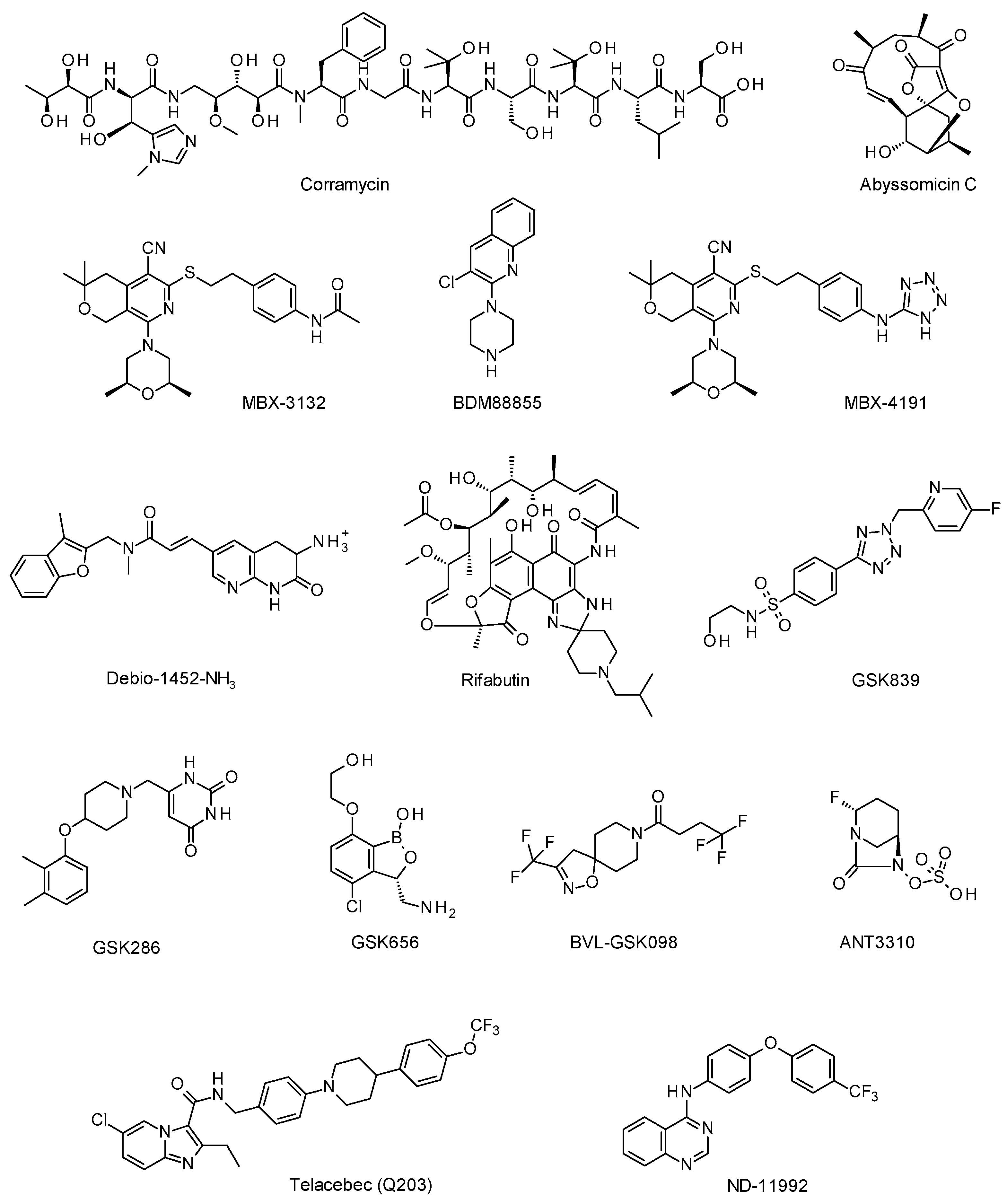
3.1.1. Corramycin: A Novel Class of Natural Antibacterials from Myxobacteria, Cédric Couturier, Ph.D. (Evotec ID, Lyon, France)
3.1.2. Antibiotic Discovery in the Abyss, Paul Race, Ph.D., Professor (University of Bristol, Bristol, United Kingdom)
3.2. Session 2: Blocking the Exit: Efflux Pump Inhibitors
3.2.1. Multidrug Efflux Pumps: Approaches on How to Get Insight in Their Structure and Function, Klaas Martinus Pos, Ph.D., Professor (Goethe University, Frankfurt, Germany)
3.2.2. Pyranopyridine EPIs as Adjunctive Therapies for MDR Enterobacteriaceae, Timothy Opperman, Ph.D. (Microbiotix, Worcester, MA, USA)
3.3. Session 3: Challenging Bacterial Membrane Permeability
3.3.1. Accumulation Rules Lead to Novel Antibiotics for Gram-Negative Bacteria, Paul Hergenrother, Ph.D., Professor (University of Illinois, Urbana-Champaign, IL, USA)
3.3.2. Rifabutin for Infusion (BV100) for the Treatment of Severe Carbapenem-Resistant Acinetobacter baumannii Infections, Glenn Dale, Ph.D. (BioVersys, Basel, Switzerland)
4. Keynote Lectures on Friday, December 10th
4.1. Session 1: Stepping Up the Fight against Tuberculosis
4.1.1. First Steps on the Road to TB Drug Candidates: Highlights and Challenges from 10 plus Years of Phenotypic Screening, Robert H. Bates Ph.D. (GSK, Tres Cantos, Spain)
4.1.2. Targeting Mycobacterial Phenotypic Variation to Potentiate Therapy and Prevent Persistence, Giulia Manina Ph.D. (Institut Pasteur, Paris, France)
4.2. Session 2: New Insights in β-Lactamase Inhibitors
4.2.1. Modulation of the Specificity of Carbapenems and Diazabicyclooctanes for Selective Activity against Mycobacterium tuberculosis, Michel Arthur, Ph.D. (Centre de Recherche des Cordeliers, Paris, France)
4.2.2. Discovery and Preclinical Development of ANT3310, a Broad-Spectrum Serine β-Lactamase Inhibitor Which Potentiates Meropenem against Carbapenem-Resistant Bacteria, David Davies, Ph.D. (Antabio, Labège, France)
4.3. Session 3: Suffocate the Bug with OXPHOS Inhibitors
Quest for Inhibitors of the Mycobacterial Respiratory Terminal Oxidases, Garrett Moraski (Montana State University, Bozeman, MT, USA) and Kevin Pethe, Ph.D., Associate Professor (Nanyang Technological University, Singapore)
5. Workshop
Author Contributions
Funding
Conflicts of Interest
References
- O’Neil, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 17 March 2022).
- 2020 Antibacterial Agents in Clinical and Preclinical Development: An Overview and Analysis n.d. Available online: https://www.who.int/publications/i/item/9789240021303 (accessed on 13 February 2022).
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.E.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541. [Google Scholar] [CrossRef] [PubMed]
- Rossiter, S.E.; Fletcher, M.H.; Wuest, W.M. Natural Products as Platforms To Overcome Antibiotic Resistance. Chem. Rev. 2017, 117, 12415. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Stennett, H.L.; Back, C.R.; Tiwari, K.; Gomez, J.O.; Challand, M.R.; Hendry, K.R.; Spencer, J.; Essex-Lopresti, A.E.; Willis, C.L.; et al. The Bristol Sponge Microbiome Collection: A Unique Repository of Deep-Sea Microorganisms and Associated Natural Products. Antibiotics 2020, 9, 509. [Google Scholar] [CrossRef]
- Back, C.R.; Stennett, H.L.; Williams, S.E.; Wang, L.; Ojeda Gomez, J.; Abdulle, O.M.; Duffy, T.; Neal, C.; Mantell, J.; Jepson, M.; et al. A New Micromonospora Strain with Antibiotic Activity Isolated from the Microbiome of a Mid-Atlantic Deep-Sea Sponge. Mar. Drugs 2021, 19, 105. [Google Scholar] [CrossRef]
- Sadaka, C.; Ellsworth, E.; Hansen, P.R.; Ewin, R.; Damborg, P.; Watts, J.L. Review on Abyssomicins: Inhibitors of the Chorismate Pathway and Folate Biosynthesis. Molecules 2018, 23, 1371. [Google Scholar] [CrossRef] [Green Version]
- Lees, N.R.; Han, L.-C.; Byrne, M.J.; Davies, J.A.; Parnell, A.E.; Moreland, P.E.J.; Stach, J.E.M.; van Der Kamp, M.W.; Willis, C.L.; Race, P.R. An Esterase-like Lyase Catalyzes Acetate Elimination in Spirotetronate/Spirotetramate Biosynthesis. Angew. Chem. 2019, 131, 2327. [Google Scholar] [CrossRef]
- Byrne, M.J.; Lees, N.R.; Han, L.C.; Van Der Kamp, M.W.; Mulholland, A.J.; Stach, J.E.M.; Willis, C.L.; Race, P.R. The Catalytic Mechanism of a Natural Diels-Alderase Revealed in Molecular Detail. J. Am. Chem. Soc. 2016, 138, 6095. [Google Scholar] [CrossRef] [Green Version]
- Alav, I.; Kobylka, J.; Kuth, M.S.; Pos, K.M.; Picard, M.; Blair, J.M.A.; Bavro, V.N. Structure, Assembly, and Function of Tripartite Efflux and Type 1 Secretion Systems in Gram-Negative Bacteria. Chem. Rev. 2021, 121, 5479. [Google Scholar] [CrossRef]
- Seeger, M.A.; Schiefner, A.; Eicher, T.; Verrey, F.; Diederichs, K.; Pos, K.M. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 2006, 313, 1295. [Google Scholar] [CrossRef] [Green Version]
- Tam, H.K.; Foong, W.E.; Oswald, C.; Herrmann, A.; Zeng, H.; Pos, K.M. Allosteric drug transport mechanism of multidrug transporter AcrB. Nat. Commun. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Sjuts, H.; Vargiu, A.V.; Kwasny, S.M.; Nguyen, S.T.; Kim, H.-S.; Ding, X.; Ornik, A.R.; Ruggerone, P.; Bowlin, T.L.; Nikaido, H.; et al. Molecular basis for inhibition of AcrB multidrug efflux pump by novel and powerful pyranopyridine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, 3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plé, C.; Tam, H.-K.; Da Cruz, A.V.; Compagne, N.; Jiménez-Castellanos, J.-C.; Müller, R.T.; Pradel, E.; Foong, W.E.; Malloci, G.; Ballée, A.; et al. Pyridylpiperazine-based allosteric inhibitors of RND-type multidrug efflux pumps. Nat. Commun. 2022, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Opperman, T.J.; Kwasny, S.M.; Kim, H.S.; Nguyen, S.T.; Houseweart, C.; D’Souza, S.; Walker, G.C.; Peet, N.P.; Nikaido, H.; Bowlin, T.L. Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother. 2014, 58, 722. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. ELife 2017, 6, e24905. [Google Scholar] [CrossRef]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371. [Google Scholar] [CrossRef]
- Richter, M.F.; Hergenrother, P.J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci. 2019, 1435, 18. [Google Scholar] [CrossRef] [Green Version]
- Geddes, E.J.; Li, Z.; Hergenrother, P.J. An LC-MS/MS assay and complementary web-based tool to quantify and predict compound accumulation in E. coli. Nat. Protoc. 2021, 16, 4833. [Google Scholar] [CrossRef]
- Richter, M.F.; Drown, B.S.; Riley, A.P.; Garcia, A.; Shirai, T.; Svec, R.L.; Hergenrother, P.J. Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299. [Google Scholar] [CrossRef] [Green Version]
- Parker, E.N.; Drown, B.; Geddes, E.J.; Lee, H.Y.; Ismail, N.; Lau, G.W.; Hergenrother, P.J. Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol. 2020, 5, 67. [Google Scholar] [CrossRef]
- Ayobami, O.; Willrich, N.; Harder, T.; Okeke, I.N.; Eckmanns, T.; Markwart, R. The incidence and prevalence of hospital-acquired (carbapenem-resistant) Acinetobacter baumannii in Europe, Eastern Mediterranean and Africa: A systematic review and meta-analysis. Emerg. Microbes Infect. 2019, 8, 1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, D.; Nielsen, T.B.; Bonomo, R.A.; Pantapalangkoor, P.; Luna, B.; Spellberg, B. Clinical and Pathophysiological Overview of Acinetobacter Infections: A Century of Challenges. Clin. Microbiol. Rev. 2017, 30, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, B.; Trebosc, V.; Lee, B.; Bakowski, M.; Ulhaq, A.; Yan, J.; Lu, P.; Cheng, J.; Nielsen, T.; Lim, J.; et al. A nutrient-limited screen unmasks rifabutin hyperactivity for extensively drug-resistant Acinetobacter baumannii. Nat. Microbiol. 2020, 5, 1134. [Google Scholar] [CrossRef] [PubMed]
- Trebosc, V.; Schellhorn, B.; Schill, J.; Lucchini, V.; Bühler, J.; Bourotte, M.; Butcher, J.J.; Gitzinger, M.; Lociuro, S.; Kemmer, C.; et al. In vitro activity of rifabutin against 293 contemporary carbapenem-resistant Acinetobacter baumannii clinical isolates and characterization of rifabutin mode of action and resistance mechanisms. J. Antimicrob. Chemother. 2020, 75, 3552. [Google Scholar] [CrossRef]
- Global Tuberculosis Report 2021; Word Health Organization: Geneva, Switzerland, 2021; Available online: https://www.who.int/publications/i/item/9789240037021 (accessed on 17 March 2022).
- Pai, M.; Kasaeva, T.; Swaminathan, S. COVID-19’s Devastating Effect on Tuberculosis Care—A Path to Recovery. N. Engl. J. Med. 2022, 5, 1134. [Google Scholar] [CrossRef]
- Abrahams, K.; Chung, C.-W.; Ghidelli-Disse, S.; Rullas, J.; Rebollo-López, M.J.; Gurcha, S.S.; Cox, J.; Mendoza, A.; Jiménez-Navarro, E.; Martínez-Martínez, M.S.; et al. Identification of KasA as the cellular target of an anti-tubercular scaffold. Nat. Commun. 2016, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- Manina, G.; Griego, A.; Singh, L.K.; McKinney, J.D.; Dhar, N. Preexisting variation in DNA damage response predicts the fate of single mycobacteria under stress. EMBO J. 2019, 38, e101876. [Google Scholar] [CrossRef]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, J.-L.; Fourgeaud, M.; Hugonnet, J.-E.; Dubost, L.; Brouard, J.-P.; Ouazzani, J.; Rice, L.B.; Gutmann, L.; Arthur, M. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J. Biol. Chem. 2005, 280, 38146. [Google Scholar] [CrossRef] [Green Version]
- Lavollay, M.; Arthur, M.; Fourgeaud, M.; Dubost, L.; Marie, A.; Veziris, N.; Blanot, D.; Gutmann, L.; Mainardi, J.-L. The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J. Bacteriol. 2008, 190, 4360. [Google Scholar] [CrossRef] [Green Version]
- Triboulet, S.; Dubée, V.; Lecoq, L.; Bougault, C.; Mainardi, J.-L.; Rice, L.B.; Ethève-Quelquejeu, M.; Gutmann, L.; Marie, A.; Dubost, L.; et al. Kinetic features of L,D-transpeptidase inactivation critical for β-lactam antibacterial activity. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triboulet, S.; Edoo, Z.; Compain, F.; Ourghanlian, C.; Dupuis, A.; Dubée, V.; Sutterlin, L.; Atze, H.; Etheve-Quelquejeu, M.; Hugonnet, J.-E.; et al. Tryptophan Fluorescence Quenching in β-Lactam-Interacting Proteins Is Modulated by the Structure of Intermediates and Final Products of the Acylation Reaction. ACS Infect. Dis. 2019, 5, 1169. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, L.; Soroka, D.; Triboulet, S.; Fonvielle, M.; Compain, F.; Dubée, V.; Mainardi, J.-L.; Hugonnet, J.-E.; Braud, E.; Arthur, M.; et al. Routes of Synthesis of Carbapenems for Optimizing Both the Inactivation of L,D-Transpeptidase LdtMt1 of Mycobacterium tuberculosis and the Stability toward Hydrolysis by β-Lactamase BlaC. J. Med. Chem. 2016, 59, 3427. [Google Scholar] [CrossRef] [PubMed]
- Edoo, Z.; Iannazzo, L.; Compain, F.; Gallay, I.L.D.L.S.; Van Tilbeurgh, H.; Fonvielle, M.; Bouchet, F.; Le Run, E.; Mainardi, J.-L.; Arthur, M.; et al. Synthesis of Avibactam Derivatives and Activity on β-Lactamases and Peptidoglycan Biosynthesis Enzymes of Mycobacteria. Chem. A Eur. J. 2018, 24, 8081. [Google Scholar] [CrossRef]
- Davies, D.T.; Leiris, S.; Zalacain, M.; Sprynski, N.; Castandet, J.; Bousquet, J.; Lozano, C.; Llanos, A.; Alibaud, L.; Vasa, S.; et al. Discovery of ANT3310, a Novel Broad-Spectrum Serine β-Lactamase Inhibitor of the Diazabicyclooctane Class, Which Strongly Potentiates Meropenem Activity against Carbapenem-Resistant Enterobacterales and Acinetobacter baumannii. J. Med. Chem. 2020, 63, 15802. [Google Scholar] [CrossRef]
- Connolly, L.E.; Edelstein, P.H.; Ramakrishnan, L. Why Is Long-Term Therapy Required to Cure Tuberculosis? PLoS Med. 2007, 4, e120. [Google Scholar] [CrossRef] [Green Version]
- Moraski, G.C.; Chang, M.; Villegas-Estrada, A.; Franzblau, S.G.; Möllmann, U.; Miller, M.J. Structure–activity relationship of new anti-tuberculosis agents derived from oxazoline and oxazole benzyl esters. Eur. J. Med. Chem. 2010, 45, 1703. [Google Scholar] [CrossRef] [Green Version]
- Moraski, G.; Markley, L.D.; Cramer, J.; Hipskind, P.A.; Boshoff, H.; Bailey, M.A.; Alling, T.; Ollinger, J.; Parish, T.; Miller, M.J.; et al. Advancement of imidazo[1,2-a]pyridines with improved pharmacokinetics and nM activity vs. Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2013, 4, 675. [Google Scholar] [CrossRef]
- Moraski, G.; Markley, L.D.; Chang, M.; Cho, S.; Franzblau, S.; Hwang, C.H.; Boshoff, H.; Miller, M.J. Generation and exploration of new classes of antitubercular agents: The optimization of oxazolines, oxazoles, thiazolines, thiazoles to imidazo[1,2-a]pyridines and isomeric 5,6-fused scaffolds. Bioorganic Med. Chem. 2012, 20, 2214. [Google Scholar] [CrossRef] [Green Version]
- Moraski, G.C.; Markley, L.D.; Hipskind, P.A.; Boshoff, H.; Cho, S.; Franzblau, S.G.; Miller, M.J. Advent of imidazo[1,2-a]pyridine-3-carboxamides with potent multi- and extended drug resistant antituberculosis activity. ACS Med. Chem. Lett. 2011, 2, 466. [Google Scholar] [CrossRef]
- Rao, S.P.S.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mak, P.A.; Rao, S.P.S.; Tan, M.P.; Lin, X.; Chyba, J.; Tay, J.; Ng, S.H.; Tan, B.H.; Cherian, J.; Duraiswamy, J.; et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157. [Google Scholar] [CrossRef] [PubMed]
- De Jager, V.R.; Dawson, R.; Van Niekerk, C.; Hutchings, J.; Kim, J.; Vanker, N.; Van Der Merwe, L.; Choi, J.; Nam, K.; Diacon, A.H. Telacebec (Q203), a New Antituberculosis Agent. N. Engl. J. Med. 2020, 382, 1280. [Google Scholar] [CrossRef] [PubMed]
- Kalia, N.P.; Hasenoehrl, E.J.; Ab Rahman, N.B.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Grüber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426. [Google Scholar] [CrossRef] [Green Version]
- Scherr, N.; Bieri, R.; Thomas, S.S.; Chauffour, A.; Kalia, N.P.; Schneide, P.; Ruf, M.-T.; Lamelas, A.; Manimekalai, M.S.S.; Grüber, G.; et al. Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nat. Commun. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.S.; Hards, K.; Engelhart, C.A.; Hasenoehrl, E.J.; Kalia, N.P.; Mackenzie, J.S.; Sviriaeva, E.; Chong, S.M.S.; Manimekalai, M.S.S.; Koh, V.H.; et al. Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis. EMBO Mol. Med. 2021, 13, e13207. [Google Scholar] [CrossRef]
- Hopfner, S.M.; Lee, B.S.; Kalia, N.P.; Miller, M.J.; Pethe, K.; Moraski, G.C. Structure guided generation of thieno[3,2-d]pyrimidin-4-amine Mycobacterium tuberculosis bd oxidase inhibitors. RSC Med. Chem. 2021, 12, 73. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antraygues, K.; Compagne, N.; Ruggieri, F.; Djaout, K.; Edoo, Z.; Eveque, M.; Faïon, L.; Gioia, B.; Tangara, S.; Vieira Da Cruz, A.; et al. On the Hunt for Next-Generation Antimicrobial Agents: An Online Symposium Organized Jointly by the French Society for Medicinal Chemistry (Société de Chimie Thérapeutique) and the French Microbiology Society (Société Française de Microbiologie) on 9–10 December 2021. Pharmaceuticals 2022, 15, 388. https://doi.org/10.3390/ph15040388
Antraygues K, Compagne N, Ruggieri F, Djaout K, Edoo Z, Eveque M, Faïon L, Gioia B, Tangara S, Vieira Da Cruz A, et al. On the Hunt for Next-Generation Antimicrobial Agents: An Online Symposium Organized Jointly by the French Society for Medicinal Chemistry (Société de Chimie Thérapeutique) and the French Microbiology Society (Société Française de Microbiologie) on 9–10 December 2021. Pharmaceuticals. 2022; 15(4):388. https://doi.org/10.3390/ph15040388
Chicago/Turabian StyleAntraygues, Kevin, Nina Compagne, Francesca Ruggieri, Kamel Djaout, Zainab Edoo, Maxime Eveque, Léo Faïon, Bruna Gioia, Salia Tangara, Anais Vieira Da Cruz, and et al. 2022. "On the Hunt for Next-Generation Antimicrobial Agents: An Online Symposium Organized Jointly by the French Society for Medicinal Chemistry (Société de Chimie Thérapeutique) and the French Microbiology Society (Société Française de Microbiologie) on 9–10 December 2021" Pharmaceuticals 15, no. 4: 388. https://doi.org/10.3390/ph15040388
APA StyleAntraygues, K., Compagne, N., Ruggieri, F., Djaout, K., Edoo, Z., Eveque, M., Faïon, L., Gioia, B., Tangara, S., Vieira Da Cruz, A., Villemagne, B., Flipo, M., Baulard, A., & Willand, N. (2022). On the Hunt for Next-Generation Antimicrobial Agents: An Online Symposium Organized Jointly by the French Society for Medicinal Chemistry (Société de Chimie Thérapeutique) and the French Microbiology Society (Société Française de Microbiologie) on 9–10 December 2021. Pharmaceuticals, 15(4), 388. https://doi.org/10.3390/ph15040388