
Metabolic Reprogramming in Melanoma: An Epigenetic Point of View

 , , , ,
, , , ,
Abstract

1. Introduction
2. Melanoma Metabolic Reprogramming
2.1. Melanoma Metabolic Plasticity: Glycolysis vs. OXPHOS
2.2. Lipid Metabolism in Melanoma
2.3. Amino Acid Metabolism in Melanoma
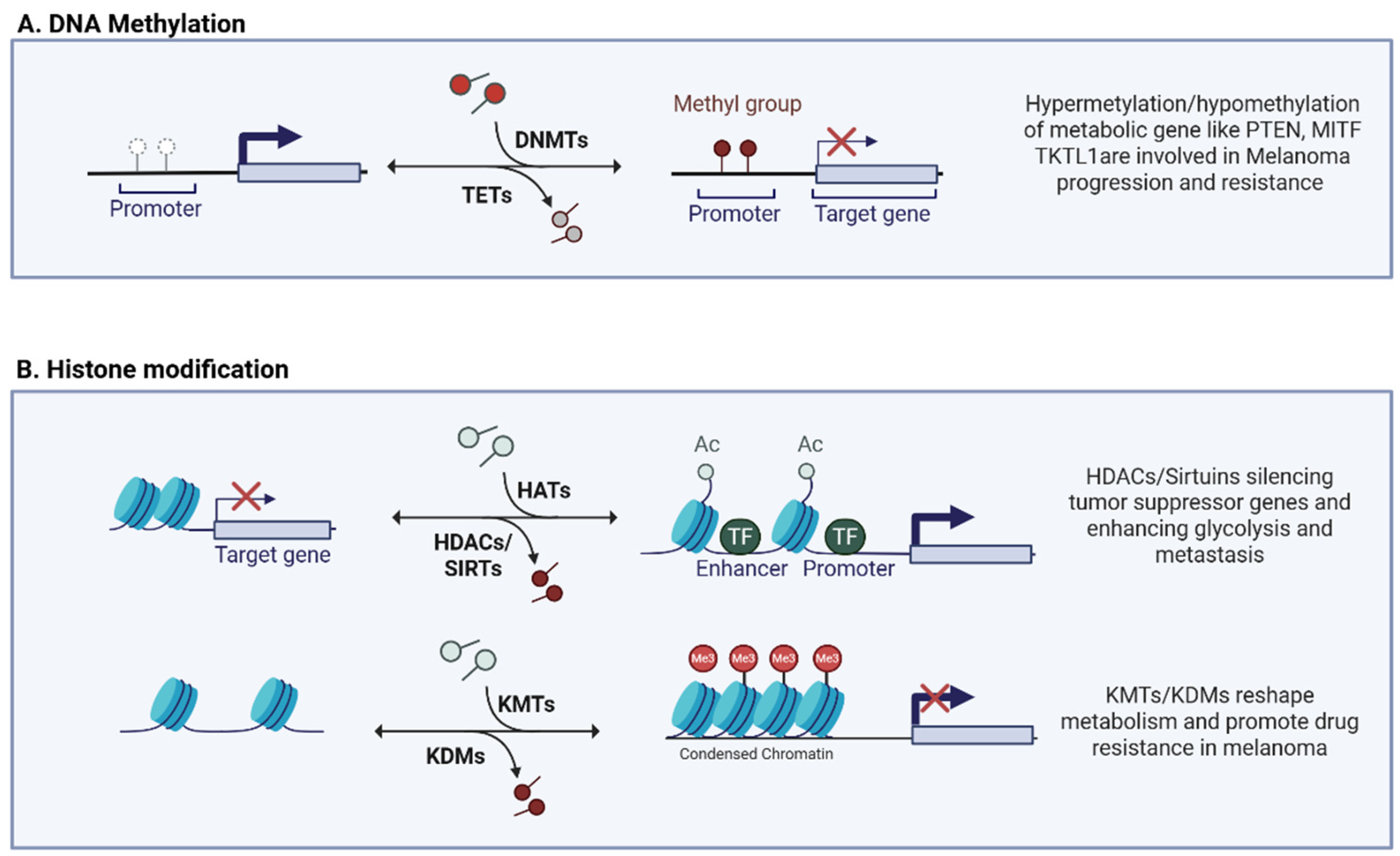
3. Epigenetic Regulation of Metabolic Plasticity
3.1. DNA Methylation
3.2. Histone Modification
3.3. Non-Coding RNAs
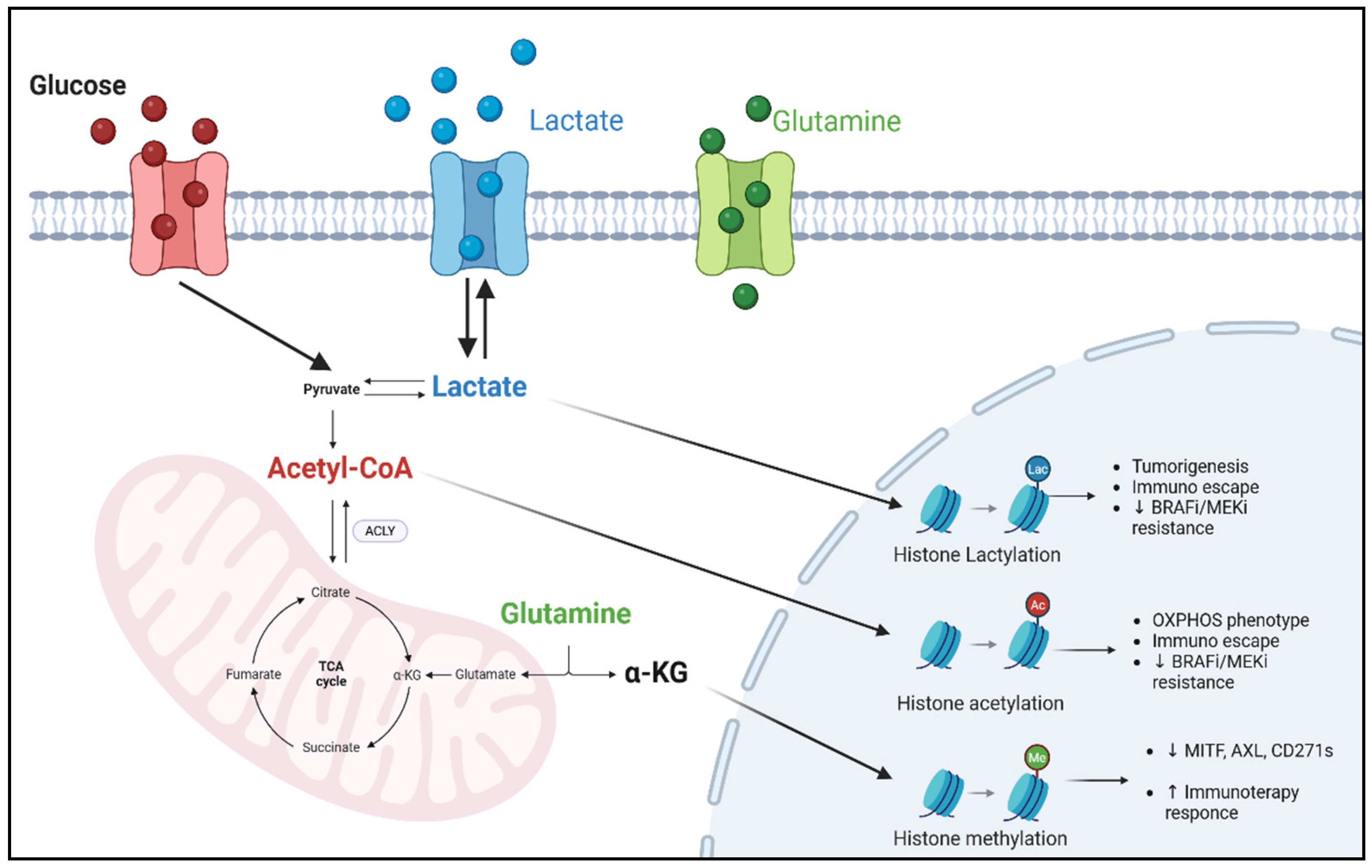
4. Metabolites as Epigenetic Modulators in Melanoma Treatment
4.1. Acetyl-CoA
4.2. α-KG and Glutamine
4.3. Lactate
5. Tumor Microenvironment and the Epigenetic–Metabolic Axis in Melanoma
6. Therapeutic Strategies and Clinical Trials Targeting the Metabolic–Epigenetic Axis in Melanoma
7. Conclusions and Future Prospective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACLY | ATP-Citrate Lyase |
| α-KG | Alpha-Ketoglutarate |
| AML | Acute Myeloid Leukemia |
| Arg1 | Arginase 1 |
| ASCT2 | Alanine–Serine–Cysteine Transporter 2 (SLC1A5) |
| BAG3 | BCL2-Associated Athanogene 3 |
| BCL2 | B-cell Leukemia/Lymphoma 2 |
| BRAFi | BRAF Inhibitor |
| circRNA | Circular RNA |
| CSC | Cancer Stem Cell |
| DNMT | DNA Methyltransferase |
| ETC | Electron Transport Chain |
| EZH2 | Enhancer of Zeste Homolog 2 |
| FAO | Fatty Acid Oxidation |
| FAs | Fatty Acids |
| FGFR1 | Fibroblast Growth Factor Receptor 1 |
| FosL1 | FOS-Like Antigen 1 |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| GSH | Reduced Glutathione |
| GSSG | Oxidized Glutathione |
| HAT | Histone Acetyltransferase |
| HDAC | Histone Deacetylase |
| HIF-1α | Hypoxia-Inducible Factor 1 Alpha |
| ICIs | Immune Checkpoint Inhibitors |
| IDH1 | Isocitrate Dehydrogenase 1 |
| KDM | Lysine Demethylase |
| KMT | Lysine Methyltransferase |
| lncRNA | Long Non-Coding RNA |
| LSD1 | Lysine-Specific Demethylase 1 |
| MAPK | Mitogen-Activated Protein Kinase |
| MCT | Monocarboxylate Transporter |
| MITF | Microphthalmia-Associated Transcription Factor |
| mTORC1 | Mechanistic Target of Rapamycin Complex 1 |
| MUFAs | Monounsaturated Fatty Acids |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| ncRNA | Non-Coding RNA |
| NO | Nitric Oxide |
| NOS | Nitric Oxide Synthase |
| OXPHOS | Oxidative Phosphorylation |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death-Ligand 1 |
| PDK | Pyruvate Dehydrogenase Kinase |
| PDH | Pyruvate Dehydrogenase |
| PER1 | Period Circadian Protein Homolog 1 |
| PHGDH | D-3-Phosphoglycerate Dehydrogenase |
| PI3K | Phosphatidylinositol 3-Kinase |
| PPP | Pentose Phosphate Pathway |
| PSPH | Phosphoserine Phosphatase |
| PSAT1 | Phosphoserine Aminotransferase 1 |
| PTEN | Phosphatase and Tensin Homolog |
| PUFAs | Polyunsaturated Fatty Acids |
| SCD1 | Stearoyl-CoA Desaturase 1 |
| SFAs | Saturated Fatty Acids |
| SIRT | Sirtuin |
| SSP | Serine Synthesis Pathway |
| STAT1/3 | Signal Transducer and Activator of Transcription 1/3 |
| TCA | Tricarboxylic Acid |
| TET | Ten-Eleven Translocation Enzyme |
| TFRC | Transferrin Receptor |
| TKTL1 | Transketolase-Like 1 |
| TME | Tumor Microenvironment |
| YTHDF2 | YTH Domain Family Protein 2 |
| CALML5 | Calmodulin-Like Protein 5 |
| CD36 | Cluster of Differentiation 36 |
| CXCL5 | C-X-C Motif Chemokine Ligand 5 |
| DMKs | Dimethyl Ketoglutarate Analogs |
| H3K18 | Histone H3 Lysine 18 Lactylation |
| H3K27ac | Histone H3 Lysine 27 Acetylation |
| IL-4 | Interleukin-4 |
| IL-6 | Interleukin-6 |
| JARID1b | Jumonji AT-Rich Interactive Domain 1B (alias KDM5B) |
| PGC1α | Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Alpha |
| SIRT1/5/6 | Sirtuins 1, 5, 6 |
References
- Mao, Y.; Xia, Z.; Xia, W.; Jiang, P. Metabolic Reprogramming, Sensing, and Cancer Therapy. Cell Rep. 2024, 43, 115064. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic Flexibility in Melanoma: A Potential Therapeutic Target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Kahn, A.M.; Perry, C.J.; Etts, K.; Kluger, H.; Sznol, M. Clinical Predictors of Survival in Patients with BRAFV600-Mutated Metastatic Melanoma Treated with Combined BRAF and MEK Inhibitors After Immune Checkpoint Inhibitors. Oncologist 2024, 29, e507–e513. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Casula, M.; Bulgarelli, J.; Pisano, M.; Piccinini, C.; Piccin, L.; Cossu, A.; Mandalà, M.; Ferrucci, P.F.; Guidoboni, M.; et al. Sequential Immunotherapy and Targeted Therapy for Metastatic BRAF V600 Mutated Melanoma: 4-Year Survival and Biomarkers Evaluation from the Phase II SECOMBIT Trial. Nat. Commun. 2024, 15, 146. [Google Scholar] [CrossRef]
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Federico, P.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic Regulation of Cell Growth and Proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Snyman, M.; Walsdorf, R.E.; Wix, S.N.; Gill, J.G. The Metabolism of Melanin Synthesis—From Melanocytes to Melanoma. Pigment Cell Melanoma Res. 2024, 37, 438–452. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The Hallmarks of Cancer Metabolism: Still Emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Zhao, J.; Jin, D.; Huang, M.; Ji, J.; Xu, X.; Wang, F.; Zhou, L.; Bao, B.; Jiang, F.; Xu, W.; et al. Glycolysis in the Tumor Microenvironment: A Driver of Cancer Progression and a Promising Therapeutic Target. Front. Cell Dev. Biol. 2024, 12, 1416472. [Google Scholar] [CrossRef]
- Zhou, D.; Duan, Z.; Li, Z.; Ge, F.; Wei, R.; Kong, L. The Significance of Glycolysis in Tumor Progression and Its Relationship with the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 1091779. [Google Scholar] [CrossRef]
- Zheng, J. Energy Metabolism of Cancer: Glycolysis Versus Oxidative Phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative Metabolic Flux Profiling of Melanoma Cell Lines: Beyond the Warburg Effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Barba, I.; Carrillo-Bosch, L.; Seoane, J. Targeting the Warburg Effect in Cancer: Where Do We Stand? Int. J. Mol. Sci. 2024, 25, 3142. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. The Warburg Metabolism Fuels Tumor Metastasis. Cancer Metastasis Rev. 2019, 38, 157–164. [Google Scholar] [CrossRef]
- Smith, L.K.; Rao, A.D.; McArthur, G.A. Targeting Metabolic Reprogramming as a Potential Therapeutic Strategy in Melanoma. Pharmacol. Res. 2016, 107, 42–47. [Google Scholar] [CrossRef]
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staibano, S.; Nappi, O.; et al. BRAF/NRAS Mutation Frequencies Among Primary Tumors and Metastases in Patients with Melanoma. J. Clin. Oncol. 2012, 30, 2522–2529. [Google Scholar] [CrossRef]
- Edlundh-Rose, E.; Egyházi, S.; Omholt, K.; Månsson-Brahme, E.; Platz, A.; Hansson, J.; Lundeberg, J. NRAS and BRAF Mutations in Melanoma Tumours in Relation to Clinical Characteristics: A Study Based on Mutation Screening by Pyrosequencing. Melanoma Res. 2006, 16, 471–478. [Google Scholar] [CrossRef]
- McGrail, K.; Granado-Martinez, P.; Esteve-Puig, R.; Garcia-Ortega, S.; Ding, Y.; Sanchez-Redondo, S.; Ferrer, B.; Hernandez-Losa, J.; Canals, F.; Manzano, A.; et al. BRAF activation by metabolic stress promotes glycolysis sensitizing NRASQ61-mutated melanomas to targeted therapy. Nat. Commun. 2022, 13, 7113. [Google Scholar] [CrossRef]
- Houles, T.; Gravel, S.P.; Lavoie, G.; Shin, S.; Savall, M.; Meant, A.; Grondin, B.; Gaboury, L.; Yoon, S.O.; St-Pierre, J.; et al. RSK Regulates PFK-2 Activity to Promote Metabolic Rewiring in Melanoma. Cancer Res. 2018, 78, 2191–2204. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J.; Puzio-Kuter, A.M. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Malekan, M.; Ebrahimzadeh, M.A.; Sheida, F. The Role of Hypoxia-Inducible Factor-1alpha and Its Signaling in Melanoma. Biomed. Pharmacother. 2021, 141, 111873. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, H.; Pino, M.S.; Zeng, M.; Shirasawa, S.; Chung, D.C. Oncogenic KRAS and BRAF Differentially Regulate Hypoxia-Inducible Factor-1α and -2α in Colon Cancer. Cancer Res. 2009, 69, 8499–8506. [Google Scholar] [CrossRef]
- Kumar, S.M.; Yu, H.; Edwards, R.; Chen, L.; Kazianis, S.; Brafford, P.; Acs, G.; Herlyn, M.; Xu, X. Mutant V600E BRAF Increases Hypoxia Inducible Factor-1α Expression in Melanoma. Cancer Res. 2007, 67, 3177–3184. [Google Scholar] [CrossRef]
- De Vitis, C.; Battaglia, A.M.; Pallocca, M.; Santamaria, G.; Mimmi, M.C.; Sacco, A.; De Nicola, F.; Gaspari, M.; Salvati, V.; Ascenzi, F.; et al. ALDOC- and ENO2-Driven Glucose Metabolism Sustains 3D Tumor Spheroids Growth Regardless of Nutrient Environmental Conditions: A Multi-Omics Analysis. J. Exp. Clin. Cancer Res. 2023, 42, 69. [Google Scholar] [CrossRef]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Singleton, K.R.; Crawford, L.; Tsui, E.; Manchester, H.E.; Maertens, O.; Liu, X.; Liberti, M.V.; Magpusao, A.N.; Stein, E.M.; Tingley, J.P.; et al. Melanoma Therapeutic Strategies that Select against Resistance by Exploiting MYC-Driven Evolutionary Convergence. Cell Rep. 2017, 21, 2796–2812. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Cell Biology. Warburg Effect and Redox Balance. Science 2011, 334, 1219–1220. [Google Scholar] [CrossRef]
- Yang, W.; Xia, Y.; Hawke, D.; Li, X.; Liang, J.; Xing, D.; Aldape, K.; Hunter, T.; Alfred Yung, W.K.; Lu, Z. PKM2 Phosphorylates Histone H3 and Promotes Gene Transcription and Tumorigenesis. Cell 2012, 150, 685–696. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, Z.; Su, J.; Li, J.; Zhao, S.; Wu, L.; Zhang, J.; He, Y.; Zhang, G.; Tao, J.; et al. Benserazide is a Novel Inhibitor Targeting PKM2 for Melanoma Treatment. Int. J. Cancer 2020, 147, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhou, Y. Crucial Role of the Pentose Phosphate Pathway in Malignant Tumors. Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [PubMed]
- Aurora, A.B.; Khivansara, V.; Leach, A.; Gill, J.G.; Martin-Sandoval, M.; Yang, C.; Kasitinon, S.Y.; Bezwada, D.; Tasdogan, A.; Gu, W.; et al. Loss of Glucose 6-Phosphate Dehydrogenase Function Increases Oxidative Stress and Glutaminolysis in Metastasizing Melanoma Cells. Proc. Natl. Acad. Sci. USA 2022, 119, e2120617119. [Google Scholar] [CrossRef]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.R.; Moore, J.A.; Bowles, K.M.; Rushworth, S.A.; Moncrieff, M.D. Mitochondrial Oxidative Phosphorylation in Cutaneous Melanoma. Br. J. Cancer 2021, 124, 115–123. [Google Scholar] [CrossRef]
- Najem, A.; Soumoy, L.; Sabbah, M.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells 2022, 11, 1157. [Google Scholar] [CrossRef]
- Ho, J.; de Moura, M.B.; Lin, Y.; Vincent, G.; Thorne, S.; Duncan, L.M.; Hui-Min, L.; Kirkwood, J.M.; Becker, D.; Van Houten, B.; et al. Importance of Glycolysis and Oxidative Phosphorylation in Advanced Melanoma. Mol. Cancer 2012, 11, 76. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting Mitochondrial Biogenesis to Overcome Drug Resistance to MAPK Inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- McArthur, G.A.; Puzanov, I.; Amaravadi, R.; Ribas, A.; Chapman, P.; Kim, K.B.; Sosman, J.A.; Lee, R.J.; Nolop, K.; Flaherty, K.T.; et al. Marked, Homogeneous, and Early [18F]Fluorodeoxyglucose-Positron Emission Tomography Responses to Vemurafenib in BRAF-Mutant Advanced Melanoma. J. Clin. Oncol. 2012, 30, 1628–1634. [Google Scholar] [CrossRef]
- Smith, L.K.; Parmenter, T.; Kleinschmidt, M.; Kusnadi, E.P.; Kang, J.; Martin, C.A.; Lau, P.; Patel, R.; Lorent, J.; Papadopoli, D.; et al. Adaptive Translational Reprogramming of Metabolism Limits the Response to Targeted Therapy in BRAFV600 Melanoma. Nat. Commun. 2022, 13, 1100. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tai, Z.; Miao, F.; Huang, H.; Zhu, Q.; Bao, L.; Chen, Z. Metabolism Heterogeneity in Melanoma Fuels Deactivation of Immunotherapy: Predict Before Protect. Front. Oncol. 2022, 12, 1046102. [Google Scholar] [CrossRef] [PubMed]
- Redondo-Muñoz, M.; Rodriguez-Baena, F.J.; Aldaz, P.; Caballé-Mestres, A.; Moncho-Amor, V.; Otaegi-Ugartemendia, M.; Carrasco-Garcia, E.; Olias-Arjona, A.; Lasheras-Otero, I.; Santamaria, E.; et al. Metabolic Rewiring Induced by Ranolazine Improves Melanoma Responses to Targeted Therapy and Immunotherapy. Nat. Metab. 2023, 5, 1544–1562. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.Y.; Yang, J.L.; Lai, R.; Zhou, Z.J.; Tang, D.; Hu, L.; Zhao, L.J. Impact of Lactate on Immune Cell Function in the Tumor Microenvironment: Mechanisms and Therapeutic Perspectives. Front. Immunol. 2025, 16, 1563303. [Google Scholar] [CrossRef]
- Terry, A.R.; Hay, N. Emerging Targets in Lipid Metabolism for Cancer Therapy. Trends Pharmacol. Sci. 2024, 45, 537–551. [Google Scholar] [CrossRef]
- Kook, E.; Kim, D.H. Elucidating the Role of Lipid-Metabolism-Related Signal Transduction and Inhibitors in Skin Cancer. Metabolites 2024, 14, 309. [Google Scholar] [CrossRef]
- Wang, R.; Yan, Q.; Liu, X.; Wu, J. Unraveling Lipid Metabolism Reprogramming for Overcoming Drug Resistance in Melanoma. Biochem. Pharmacol. 2024, 223, 116122. [Google Scholar] [CrossRef]
- Kuna, R.S.; Kumar, A.; Wessendorf-Rodriguez, K.A.; Galvez, H.; Green, C.R.; McGregor, G.H.; Cordes, T.; Shaw, R.J.; Svensson, R.U.; Metallo, C.M. Inter-Organelle Cross-Talk Supports Acetyl-Coenzyme A Homeostasis and Lipogenesis Under Metabolic Stress. Sci. Adv. 2023, 9, eadf0138. [Google Scholar] [CrossRef]
- Izzo, L.T.; Trefely, S.; Demetriadou, C.; Drummond, J.M.; Mizukami, T.; Kuprasertkul, N.; Farria, A.T.; Nguyen, P.T.T.; Murali, N.; Reich, L.; et al. Acetylcarnitine Shuttling Links Mitochondrial Metabolism to Histone Acetylation and Lipogenesis. Sci. Adv. 2023, 9, eadf0115. [Google Scholar] [CrossRef]
- Guo, W.; Ma, J.; Yang, Y.; Guo, S.; Zhang, W.; Zhao, T.; Yi, X.; Wang, H.; Wang, S.; Liu, Y.; et al. ATP-Citrate Lyase Epigenetically Potentiates Oxidative Phosphorylation to Promote Melanoma Growth and Adaptive Resistance to MAPK Inhibition. Clin. Cancer Res. 2020, 26, 2725–2739. [Google Scholar] [CrossRef]
- Tian, Y.; Ma, J.; Wang, H.; Yi, X.; Wang, H.; Zhang, H.; Guo, S.; Yang, Y.; Zhang, B.; Du, J.; et al. BCAT2 Promotes Melanoma Progression by Activating Lipogenesis via the Epigenetic Regulation of FASN and ACLY Expressions. Cell. Mol. Life Sci. 2023, 80, 315. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yi, X.; Wang, X.; Yang, Y.; Zhang, H.; Wang, H.; Chen, J.; Zhang, B.; Guo, S.; Wu, L.; et al. Nucleo-Cytosolic Acetyl-CoA Drives Tumor Immune Evasion by Regulating PD-L1 in Melanoma. Cell Rep. 2024, 43, 115015. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, F.; De Vitis, C.; Maugeri-Saccà, M.; Napoli, C.; Ciliberto, G.; Mancini, R. SCD1, Autophagy and Cancer: Implications for Therapy. J. Exp. Clin. Cancer Res. 2021, 40, 265. [Google Scholar] [CrossRef] [PubMed]
- Vivas-García, Y.; Falletta, P.; Liebing, J.; Louphrasitthiphol, P.; Feng, Y.; Chauhan, J.; Scott, D.A.; Glodde, N.; Chocarro-Calvo, A.; Bonham, S.; et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol. Cell 2020, 77, 120–137.e9. [Google Scholar] [CrossRef]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-Desaturase 1 Regulates Lung Cancer Stemness via Stabilization and Nuclear Localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA Desaturase 1 Reverts BRAF and MEK Inhibition-Induced Selection of Cancer Stem Cells in BRAF-Mutated Melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 318. [Google Scholar] [CrossRef]
- Aloia, A.; Müllhaupt, D.; Chabbert, C.D.; Eberhart, T.; Flückiger-Mangual, S.; Vukolic, A.; Eichhoff, O.; Irmisch, A.; Alexander, L.T.; Scibona, E.; et al. A Fatty Acid Oxidation-dependent Metabolic Shift Regulates the Adaptation of BRAF-mutated Melanoma to MAPK Inhibitors. Clin. Cancer Res. 2019, 25, 6852–6867. [Google Scholar] [CrossRef]
- Wasinger, C.; Hofer, A.; Spadiut, O.; Hohenegger, M. Amino Acid Signature in Human Melanoma Cell Lines from Different Disease Stages. Sci. Rep. 2018, 8, 6245. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting Glutamine Metabolism as a Therapeutic Strategy for Cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Luo, M.; Wu, L.; Zhang, K.; Wang, H.; Zhang, T.; Gutierrez, L.; O’Connell, D.; Zhang, P.; Li, Y.; Gao, T.; et al. miR-137 Regulates Ferroptosis by Targeting Glutamine Transporter SLC1A5 in Melanoma. Cell Death Differ. 2018, 25, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Beaumont, K.A.; Otte, N.J.; Font, J.; Bailey, C.G.; van Geldermalsen, M.; Sharp, D.M.; Tiffen, J.C.; Ryan, R.M.; Jormakka, M.; et al. Targeting Glutamine Transport to Suppress Melanoma Cell Growth. Int. J. Cancer 2014, 135, 1060–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, S.; Gabisi, A.M., Jr.; Er, J.A.; Pope, A.; Burstein, G.; Schardon, C.L.; Cardounel, A.J.; Ekmekcioglu, S.; Fast, W. Developing an Irreversible Inhibitor of Human DDAH-1, an Enzyme Upregulated in Melanoma. ChemMedChem 2014, 9, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.K.; Frankel, A.E.; Feun, L.G.; Ekmekcioglu, S.; Kim, K.B. Arginine Deprivation Therapy for Malignant Melanoma. Clin. Pharmacol. 2013, 5, 11–19. [Google Scholar] [CrossRef]
- Kim, S.H.; Roszik, J.; Grimm, E.A.; Ekmekcioglu, S. Impact of l-Arginine Metabolism on Immune Response and Anticancer Immunotherapy. Front. Oncol. 2018, 8, 67. [Google Scholar] [CrossRef]
- Sullivan, M.R.; Mattaini, K.R.; Dennstedt, E.A.; Nguyen, A.A.; Sivanand, S.; Reilly, M.F.; Meeth, K.; Muir, A.; Darnell, A.M.; Bosenberg, M.W.; et al. Increased Serine Synthesis Provides an Advantage for Tumors Arising in Tissues Where Serine Levels Are Limiting. Cell Metab. 2019, 29, 1410–1421.e4. [Google Scholar] [CrossRef]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and Glycine Metabolism in Cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef]
- Nguyen, M.Q.; Teh, J.L.F.; Purwin, T.J.; Chervoneva, I.; Davies, M.A.; Nathanson, K.L.; Cheng, P.F.; Levesque, M.P.; Dummer, R.; Aplin, A.E. Targeting PHGDH Upregulation Reduces Glutathione Levels and Resensitizes Resistant NRAS-Mutant Melanoma to MAPK Kinase Inhibition. J. Investig. Dermatol. 2020, 140, 2242–2252.e7. [Google Scholar] [CrossRef]
- Jasani, N.; Xu, X.; Posorske, B.; Kim, Y.; Wang, K.; Vera, O.; Tsai, K.Y.; DeNicola, G.M.; Karreth, F.A. PHGDH Induction by MAPK Is Essential for Melanoma Formation and Creates an Actionable Metabolic Vulnerability. Cancer Res. 2025, 85, 314–328. [Google Scholar] [CrossRef]
- Karami Fath, M.; Azargoonjahromi, A.; Soofi, A.; Almasi, F.; Hosseinzadeh, S.; Khalili, S.; Sheikhi, K.; Ferdousmakan, S.; Owrangi, S.; Fahimi, M.; et al. Current Understanding of Epigenetics Role in Melanoma Treatment and Resistance. Cancer Cell Int. 2022, 22, 313. [Google Scholar] [CrossRef]
- Guo, W.-N.; Li, C.-Y. Linking Cellular Metabolism to Epigenetics in Melanoma. Int. J. Dermatol. Venereol. 2021, 4, 168–173. [Google Scholar] [CrossRef]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Kinnaird, A.; Zhao, S.; Wellen, K.E.; Michelakis, E.D. Metabolic Control of Epigenetics in Cancer. Nat. Rev. Cancer 2016, 16, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.; Robertson, K.D. DNA methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes Cancer 2011, 2, 607–617. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends Genet. 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Wong, C.C.; Qian, Y.; Yu, J. Interplay Between Epigenetics and Metabolism in Oncogenesis: Mechanisms and Therapeutic Approaches. Oncogene 2017, 36, 3359–3374. [Google Scholar] [CrossRef]
- Aleotti, V.; Catoni, C.; Poggiana, C.; Rosato, A.; Facchinetti, A.; Scaini, M.C. Methylation Markers in Cutaneous Melanoma: Unravelling the Potential Utility of Their Tracking by Liquid Biopsy. Cancers 2021, 13, 6217. [Google Scholar] [CrossRef]
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA Methylation in Melanoma: Biomarker and Therapeutic Opportunities. Clin. Epigenet. 2017, 9, 34. [Google Scholar] [CrossRef]
- de Araújo, É.S.; Pramio, D.T.; Kashiwabara, A.Y.; Pennacchi, P.C.; Maria-Engler, S.S.; Achatz, M.I.; Campos, A.H.; Duprat, J.P.; Rosenberg, C.; Carraro, D.M.; et al. DNA Methylation Levels of Melanoma Risk Genes Are Associated with Clinical Characteristics of Melanoma Patients. BioMed Res. Int. 2015, 2015, 376423. [Google Scholar] [CrossRef]
- Roh, M.R.; Gupta, S.; Park, K.H.; Chung, K.Y.; Lauss, M.; Flaherty, K.T.; Jönsson, G.; Rha, S.Y.; Tsao, H. Promoter Methylation of PTEN Is a Significant Prognostic Factor in Melanoma Survival. J. Investig. Dermatol. 2016, 136, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Lahtz, C.; Stranzenbach, R.; Fiedler, E.; Helmbold, P.; Dammann, R.H. Methylation of PTEN as a Prognostic Factor in Malignant Melanoma of the Skin. J. Investig. Dermatol. 2010, 130, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Giannitti, G.; Marchesi, S.; Limonta, P. The PI3K/Akt Pathway and Glucose Metabolism: A Dangerous Liaison in Cancer. Int. J. Biol. Sci. 2024, 20, 3113–3125. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, S.; Ye, Z.; Zheng, Y.; Zheng, Z.; Liu, X.; Zhou, X. Oncogenic DNA Methyltransferase 1 Activates the PI3K/AKT/mTOR Signalling by Blocking the Binding of HSPB8 and BAG3 in Melanoma. Epigenetics 2023, 18, 2239607. [Google Scholar] [CrossRef]
- Lauss, M.; Haq, R.; Cirenajwis, H.; Phung, B.; Harbst, K.; Staaf, J.; Rosengren, F.; Holm, K.; Aine, M.; Jirstrom, K.; et al. Genome-Wide DNA Methylation Analysis in Melanoma Reveals the Importance of CpG Methylation in MITF Regulation. J. Investig. Dermatol. 2015, 135, 1820–1828. [Google Scholar] [CrossRef]
- Falletta, P.; Goding, C.R.; Vivas-García, Y. Connecting Metabolic Rewiring with Phenotype Switching in Melanoma. Front. Cell Dev. Biol. 2022, 10, 930250. [Google Scholar] [CrossRef]
- Zhu, X.; Xuan, Z.; Chen, J.; Li, Z.; Zheng, S.; Song, P. How DNA Methylation Affects the Warburg Effect. Int. J. Biol. Sci. 2020, 16, 2029–2041. [Google Scholar] [CrossRef]
- Jayachandran, A.; Lo, P.H.; Chueh, A.C.; Prithviraj, P.; Molania, R.; Davalos-Salas, M.; Anaka, M.; Walkiewicz, M.; Cebon, J.; Behren, A. Transketolase-Like 1 Ectopic Expression is Associated with DNA Hypomethylation and Induces the Warburg Effect in Melanoma Cells. BMC Cancer 2016, 16, 134. [Google Scholar] [CrossRef]
- Zhou, S.; Zeng, H.; Huang, J.; Lei, L.; Tong, X.; Li, S.; Zhou, Y.; Guo, H.; Khan, M.; Luo, L.; et al. Epigenetic Regulation of Melanogenesis. Ageing Res. Rev. 2021, 69, 101349. [Google Scholar] [CrossRef]
- Sutopo, N.C.; Kim, J.H.; Cho, J.Y. Role of Histone Methylation in Skin Cancers: Histone Methylation-Modifying Enzymes as a New Class of Targets for Skin Cancer Treatment. Biochim. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 188865. [Google Scholar] [CrossRef]
- Peserico, A.; Simone, C. Physical and Functional HAT/HDAC Interplay Regulates Protein Acetylation Balance. J. Biomed. Biotechnol. 2011, 2011, 371832. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.J.; Tiffen, J.C.; Hersey, P. Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers 2015, 7, 1959–1982. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Cho, H.; Yoo, J.; Kim, G.W.; Jeon, Y.H.; Lee, S.W.; Kwon, S.H. HDAC8 Deacetylates HIF-1α and Enhances Its Protein Stability to Promote Tumor Growth and Migration in Melanoma. Cancers 2023, 15, 1123. [Google Scholar] [CrossRef] [PubMed]
- Leotta, C.G.; Barbaraci, C.; Fiorito, J.; Coco, A.; di Giacomo, V.; Amata, E.; Marrazzo, A.; Pitari, G.M. HDAC/σ1R Dual-Ligand as a Targeted Melanoma Therapeutic. Pharmaceuticals 2025, 18, 179. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Ndiaye, M.A.; Singh, C.K.; Chhabra, G.; Huang, W.; Ahmad, N. SIRT6 Histone Deacetylase Functions as a Potential Oncogene in Human Melanoma. Genes Cancer 2017, 8, 701–712. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in Skin and Skin Cancers. Skin Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef]
- Maitituoheti, M.; Keung, E.Z.; Tang, M.; Yan, L.; Alam, H.; Han, G.; Singh, A.K.; Raman, A.T.; Terranova, C.; Sarkar, S.; et al. Enhancer Reprogramming Confers Dependence on Glycolysis and IGF Signaling in KMT2D Mutant Melanoma. Cell Rep. 2020, 33, 108293. [Google Scholar] [CrossRef]
- Murugesan, N.; Maitituoheti, M. KMT2D Deficiency Confers a Therapeutic Vulnerability to Glycolytic and IGFR Inhibitors in Melanoma. Mol. Cell. Oncol. 2021, 8, 1984827. [Google Scholar] [CrossRef]
- Vogel, F.C.E.; Bordag, N.; Zügner, E.; Trajkovic-Arsic, M.; Chauvistré, H.; Shannan, B.; Váraljai, R.; Horn, S.; Magnes, C.; Thomas Siveke, J.; et al. Targeting the H3K4 Demethylase KDM5B Reprograms the Metabolome and Phenotype of Melanoma Cells. J. Investig. Dermatol. 2019, 139, 2506–2516.e10. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-Coding RNA: What is Functional and What is Junk? Front. Genet. 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Grafanaki, K.; Grammatikakis, I.; Ghosh, A.; Gopalan, V.; Olgun, G.; Liu, H.; Kyriakopoulos, G.C.; Skeparnias, I.; Georgiou, S.; Stathopoulos, C.; et al. Noncoding RNA Circuitry in Melanoma Onset, Plasticity, and Therapeutic Response. Pharmacol. Ther. 2023, 248, 108466. [Google Scholar] [CrossRef] [PubMed]
- Luan, W.; Zhou, Z.; Ni, X.; Xia, Y.; Wang, J.; Yan, Y.; Xu, B. Long Non-Coding RNA H19 Promotes Glucose Metabolism and Cell Growth in Malignant Melanoma via miR-106a-5p/E2F3 Axis. J. Cancer Res. Clin. Oncol. 2018, 144, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, D.; Xiao, M.; Zhu, Y.; Zhou, J.; Cao, K. Long Noncoding RNA LINC00518 Induces Radioresistance by Regulating Glycolysis Through an miR-33a-3p/HIF-1α Negative Feedback Loop in Melanoma. Cell Death Dis. 2021, 12, 245. [Google Scholar] [CrossRef]
- Ding, Z.; Yang, J.; Wu, B.; Wu, Y.; Guo, F. Long Non-Coding RNA CCHE1 Modulates LDHA-Mediated Glycolysis and Confers Chemoresistance to Melanoma Cells. Cancer Metab. 2023, 11, 10. [Google Scholar] [CrossRef]
- Jin, C.; Dong, D.; Yang, Z.; Xia, R.; Tao, S.; Piao, M. CircMYC Regulates Glycolysis and Cell Proliferation in Melanoma. Cell Biochem. Biophys. 2020, 78, 77–88. [Google Scholar] [CrossRef]
- Zhou, J.; Xu, D.; Xie, H.; Tang, J.; Liu, R.; Li, J.; Wang, S.; Chen, X.; Su, J.; Zhou, X.; et al. miR-33a Functions as a Tumor Suppressor in Melanoma by Targeting HIF-1α. Cancer Biol. Ther. 2015, 16, 846–855. [Google Scholar] [CrossRef]
- Cao, K.; Li, J.; Chen, J.; Qian, L.; Wang, A.; Chen, X.; Xiong, W.; Tang, J.; Tang, S.; Chen, Y.; et al. microRNA-33a-5p Increases Radiosensitivity by Inhibiting Glycolysis in Melanoma. Oncotarget 2017, 8, 83660–83672. [Google Scholar] [CrossRef]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Gambi, G.; Mengus, G.; Davidson, G.; Demesmaeker, E.; Cuomo, A.; Bonaldi, T.; Katopodi, V.; Malouf, G.G.; Leucci, E.; Davidson, I. The LncRNA LENOX Interacts with RAP2C to Regulate Metabolism and Promote Resistance to MAPK Inhibition in Melanoma. Cancer Res. 2022, 82, 4555–4570. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Esmaeili, M.; Taheri, M. H19 lncRNA: Roles in Tumorigenesis. Biomed. Pharmacother. 2020, 123, 109774. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The Evolving Metabolic Landscape of Chromatin Biology and Epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, L.L.; Xiong, Y.; Ye, D. Metabolite Regulation of Epigenetics in Cancer. Cell Rep. 2024, 43, 114815. [Google Scholar] [CrossRef]
- Xia, S.; Lin, R.; Jin, L.; Zhao, L.; Kang, H.B.; Pan, Y.; Liu, S.; Qian, G.; Qian, Z.; Konstantakou, E.; et al. Prevention of Dietary-Fat-Fueled Ketogenesis Attenuates BRAF V600E Tumor Growth. Cell Metab. 2017, 25, 358–373. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Huang, H.; Dai, L.; Liu, X.; Zhao, Y.; Locasale, J.W. The Rate of Glycolysis Quantitatively Mediates Specific Histone Acetylation Sites. Cancer Metab. 2015, 3, 10. [Google Scholar] [CrossRef]
- He, W.; Li, Q.; Li, X. Acetyl-CoA Regulates Lipid Metabolism and Histone Acetylation Modification in Cancer. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188837. [Google Scholar] [CrossRef]
- Chen, G.; Bao, B.; Cheng, Y.; Tian, M.; Song, J.; Zheng, L.; Tong, Q. Acetyl-CoA Metabolism as a Therapeutic Target for Cancer. Biomed. Pharmacother. 2023, 168, 115741. [Google Scholar] [CrossRef]
- Hunt, E.G.; Hurst, K.E.; Riesenberg, B.P.; Kennedy, A.S.; Gandy, E.J.; Andrews, A.M.; Del Mar Alicea Pauneto, C.; Ball, L.E.; Wallace, E.D.; Gao, P.; et al. Acetyl-CoA Carboxylase Obstructs CD8+ T Cell Lipid Utilization in the Tumor Microenvironment. Cell Metab. 2024, 36, 969–983.e10. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Lv, H.; Xing, F.; Sun, X.; Ma, Y.; Wu, L.; Lv, G.; Zong, Q.; Wang, L.; Wu, Z.; et al. Inhibition of ACLY Overcomes Cancer Immunotherapy Resistance via Polyunsaturated Fatty Acids Peroxidation and cGAS-STING Activation. Sci. Adv. 2023, 9, eadi2465. [Google Scholar] [CrossRef] [PubMed]
- Naeini, S.H.; Mavaddatiyan, L.; Kalkhoran, Z.R.; Taherkhani, S.; Talkhabi, M. Alpha-Ketoglutarate as a Potent Regulator for Lifespan and Healthspan: EVIDENCES and Perspectives. Exp. Gerontol. 2023, 175, 112154. [Google Scholar] [CrossRef]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-Hydroxyglutarate is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Lian, C.G.; Xu, Y.; Ceol, C.; Wu, F.; Larson, A.; Dresser, K.; Xu, W.; Tan, L.; Hu, Y.; Zhan, Q.; et al. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell 2012, 150, 1135–1146. [Google Scholar] [CrossRef]
- Ishak Gabra, M.B.; Yang, Y.; Li, H.; Senapati, P.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Zhang, L.; Doan, L.T.; Xu, X.; et al. Dietary Glutamine Supplementation Suppresses Epigenetically-Activated Oncogenic Pathways to Inhibit Melanoma Tumour Growth. Nat. Commun. 2020, 11, 3326. [Google Scholar] [CrossRef]
- Wang, B.; Pei, J.; Xu, S.; Liu, J.; Yu, J. A Glutamine Tug-of-War Between Cancer and Immune Cells: Recent Advances in Unraveling the Ongoing Battle. J. Exp. Clin. Cancer Res. 2024, 43, 74. [Google Scholar] [CrossRef]
- Ma, G.; Zhang, Z.; Li, P.; Zhang, Z.; Zeng, M.; Liang, Z.; Li, D.; Wang, L.; Chen, Y.; Liang, Y.; et al. Reprogramming of Glutamine Metabolism and Its Impact on Immune Response in the Tumor Microenvironment. Cell Commun. Signal. 2022, 20, 114. [Google Scholar] [CrossRef]
- Liu, N.; Zhang, J.; Yan, M.; Chen, L.; Wu, J.; Tao, Q.; Yan, B.; Chen, X.; Peng, C. Supplementation with α-Ketoglutarate Improved the Efficacy of anti-PD1 Melanoma Treatment Through Epigenetic Modulation of PD-L1. Cell Death Dis. 2023, 14, 170. [Google Scholar] [CrossRef]
- Liang, L.; Kuang, X.; He, Y.; Zhu, L.; Lau, P.; Li, X.; Luo, D.; Gong, L.; Zhou, W.; Zhang, F.; et al. Alterations in PD-L1 Succinylation Shape Anti-Tumor Immune Responses in Melanoma. Nat. Genet. 2025, 57, 680–693. [Google Scholar] [CrossRef]
- Yu, X.; Yang, J.; Xu, J.; Pan, H.; Wang, W.; Yu, X.; Shi, S. Histone Lactylation: From Tumor Lactate Metabolism to Epigenetic Regulation. Int. J. Biol. Sci. 2024, 20, 1833–1854. [Google Scholar] [CrossRef] [PubMed]
- Merkuri, F.; Rothstein, M.; Simoes-Costa, M. Histone Lactylation Couples Cellular Metabolism with Developmental Gene Regulatory Networks. Nat. Commun. 2024, 15, 90. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Ye, Z.; Li, Z.; Jing, D.S.; Fan, G.X.; Liu, M.Q.; Zhuo, Q.F.; Ji, S.R.; Yu, X.J.; Xu, X.W.; et al. Lactate-Induced Protein Lactylation: A Bridge Between Epigenetics and Metabolic Reprogramming in Cancer. Cell Prolif. 2023, 56, e13478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic Regulation of Gene Expression by Histone Lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Longhitano, L.; Giallongo, S.; Orlando, L.; Broggi, G.; Longo, A.; Russo, A.; Caltabiano, R.; Giallongo, C.; Barbagallo, I.; Di Rosa, M.; et al. Lactate Rewrites the Metabolic Reprogramming of Uveal Melanoma Cells and Induces Quiescence Phenotype. Int. J. Mol. Sci. 2022, 24, 24. [Google Scholar] [CrossRef]
- Yu, J.; Chai, P.; Xie, M.; Ge, S.; Ruan, J.; Fan, X.; Jia, R. Histone Lactylation Drives Oncogenesis by Facilitating m6A Reader Protein YTHDF2 Expression in Ocular Melanoma. Genome Biol. 2021, 22, 85. [Google Scholar] [CrossRef]
- Li, A.; Gong, Z.; Long, Y.; Li, Y.; Liu, C.; Lu, X.; Li, Q.; He, X.; Lu, H.; Wu, K.; et al. Lactylation of LSD1 is an Acquired Epigenetic Vulnerability of BRAFi/MEKi-Resistant Melanoma. Dev. Cell, 2025; in press. [Google Scholar] [CrossRef]
- Feng, H.; Chen, W.; Zhang, C. Identification of Lactylation Gene CALML5 and Its Correlated lncRNAs in Cutaneous Melanoma by Machine Learning. Medicine 2023, 102, e35999. [Google Scholar] [CrossRef]
- Romano, V.; Belviso, I.; Venuta, A.; Ruocco, M.R.; Masone, S.; Aliotta, F.; Fiume, G.; Montagnani, S.; Avagliano, A.; Arcucci, A. Influence of Tumor Microenvironment and Fibroblast Population Plasticity on Melanoma Growth, Therapy Resistance and Immunoescape. Int. J. Mol. Sci. 2021, 22, 5283. [Google Scholar] [CrossRef]
- Falcone, I.; Conciatori, F.; Bazzichetto, C.; Ferretti, G.; Cognetti, F.; Ciuffreda, L.; Milella, M. Tumor Microenvironment: Implications in Melanoma Resistance to Targeted Therapy and Immunotherapy. Cancers 2020, 12, 2870. [Google Scholar] [CrossRef]
- Gao, Y.; Siyu, Z.; Zhang, X.; Du, Y.; Ni, T.; Hao, S. Crosstalk Between Metabolic and Epigenetic Modifications During Cell Carcinogenesis. iScience 2024, 27, 111359. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Han, Y.; Chen, Y.; Shen, H.; Zeng, S.; Cai, C. Tumor Metabolic Regulators: Key Drivers of Metabolic Reprogramming and the promising targets in cancer therapy. Mol. Cancer 2025, 24, 7. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Osborn, G.; Willsmore, Z.; Chew, M.W.; Jakubow, S.; Fitzpatrick, A.; Wu, Y.; Sinha, K.; Lloyd-Hughes, H.; Geh, J.L.C.; et al. Tumor Associated Macrophages as Key Contributors and Targets in Current and Future Therapies for Melanoma. Expert Rev. Clin. Immunol. 2024, 20, 895–911. [Google Scholar] [CrossRef] [PubMed]
- Noe, J.T.; Rendon, B.E.; Geller, A.E.; Conroy, L.R.; Morrissey, S.M.; Young, L.E.A.; Bruntz, R.C.; Kim, E.J.; Wise-Mitchell, A.; Barbosa de Souza Rizzo, M.; et al. Lactate Supports a Metabolic-Epigenetic Link in Macrophage Polarization. Sci. Adv. 2021, 7, eabi8602. [Google Scholar] [CrossRef]
- Saw, P.E.; Chen, J.; Song, E. Targeting CAFs to Overcome Anticancer Therapeutic Resistance. Trends Cancer 2022, 8, 527–555. [Google Scholar] [CrossRef]
- Kehrberg, R.J.; Bhyravbhatla, N.; Batra, S.K.; Kumar, S. Epigenetic Regulation of Cancer-Associated Fibroblast Heterogeneity. Biochim. Biophys. Acta (BBA) Rev. Cancer 2023, 1878, 188901. [Google Scholar] [CrossRef]
- Zhou, Q.; Jin, X.; Zhao, Y.; Wang, Y.; Tao, M.; Cao, Y.; Yin, X. Melanoma-Associated Fibroblasts in Tumor-Promotion Flammation and Antitumor Immunity: Novel Mechanisms and Potential Immunotherapeutic Strategies. Hum. Mol. Genet. 2024, 33, 1186–1193. [Google Scholar] [CrossRef]
- Clinicaltrials.Gov. Available online: https://clinicaltrials.gov/ (accessed on 3 June 2025).
- Noviello, T.M.R.; Di Giacomo, A.M.; Caruso, F.P.; Covre, A.; Mortarini, R.; Scala, G.; Costa, M.C.; Coral, S.; Fridman, W.H.; Sautès-Fridman, C.; et al. Guadecitabine Plus Ipilimumab in Unresectable Melanoma: Five-Year Follow-Up and Integrated Multi-Omic Analysis in the Phase 1b NIBIT-M4 trial. Nat. Commun. 2023, 14, 5914. [Google Scholar] [CrossRef]
- Gouda, M.A.; Voss, M.H.; Tawbi, H.; Gordon, M.; Tykodi, S.S.; Lam, E.T.; Vaishampayan, U.; Tannir, N.M.; Chaves, J.; Nikolinakos, P.; et al. A Phase I/II Study of the Safety and Efficacy of Telaglenastat (CB-839) in Combination with Nivolumab in Patients with Metastatic Melanoma, Renal Cell Carcinoma, and Non-Small-Cell Lung Cancer. ESMO Open 2025, 10, 104536. [Google Scholar] [CrossRef]
- Chapman, P.B.; Klang, M.; Postow, M.A.; Shoushtari, A.N.; Sullivan, R.J.; Wolchok, J.D.; Merghoub, T.; Budhu, S.; Wong, P.; Callahan, M.K.; et al. Phase Ib Trial of Phenformin in Patients with V600-mutated Melanoma Receiving Dabrafenib and Trametinib. Cancer Res. Commun. 2023, 3, 2447–2454. [Google Scholar] [CrossRef]
- McNeillis, R.; Greystoke, A.; Walton, J.; Bacon, C.; Keun, H.; Siskos, A.; Petrides, G.; Leech, N.; Jenkinson, F.; Bowron, A.; et al. A Case of Malignant Hyperlactaemic Acidosis Appearing Upon Treatment with the Mono-Carboxylase Transporter 1 Inhibitor AZD3965. Br. J. Cancer 2020, 122, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Fakih, M.; Schneider, C.; Chiorean, E.G.; Bendell, J.; Kristeleit, R.; Kurzrock, R.; Blagden, S.P.; Brana, I.; Goff, L.W.; et al. Phase I/II Sequencing Study of Azacitidine, Epacadostat, and Pembrolizumab in Advanced Solid Tumors. Br. J. Cancer 2023, 128, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, B.J.; van Baren, N.; Baurain, J.-F. Is There a Clinical Future for IDO1 Inhibitors After the Failure of Epacadostat in Melanoma? Annu. Rev. Cancer Biol. 2020, 4, 241–256. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, Z.; Huo, X.; Gong, Y.; Li, C.; Huang, J.; Wang, Y.; Feng, H.; Ma, X.; Jiang, C.; et al. Dysregulated Lipid Metabolism Blunts the Sensitivity of Cancer Cells to EZH2 Inhibitor. EBioMedicine 2022, 77, 103872. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Gu, X.; Jia, R.; Ge, S.; Chai, P.; Zhuang, A.; Fan, X. Crosstalk Between Metabolic Reprogramming and Epigenetics in Cancer: Updates on Mechanisms and Therapeutic Opportunities. Cancer Commun. 2022, 42, 1049–1082. [Google Scholar] [CrossRef]
- Zakharia, Y.; Monga, V.; Swami, U.; Bossler, A.D.; Freesmeier, M.; Frees, M.; Khan, M.; Frydenlund, N.; Srikantha, R.; Vanneste, M.; et al. Targeting Epigenetics for Treatment of BRAF Mutated Metastatic Melanoma with Decitabine in Combination with Vemurafenib: A Phase lb Study. Oncotarget 2017, 8, 89182–89193. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Jänne, P.A.; Opyrchal, M.; Hafez, N.; Raez, L.E.; Gabrilovich, D.I.; Wang, F.; Trepel, J.B.; Lee, M.J.; Yuno, A.; et al. Entinostat Plus Pembrolizumab in Patients with Metastatic NSCLC Previously Treated with Anti-PD-(L)1 Therapy. Clin. Cancer Res. 2021, 27, 1019–1028. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat Plus Pembrolizumab Versus Placebo Plus Pembrolizumab in Patients with Unresectable or Metastatic Melanoma (ECHO-301/KEYNOTE-252): A Phase 3, Randomised, Double-Blind Study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| ncRNA | Effect | Citation |
|---|---|---|
| H19 | Upregulates glycolysis via HIF-1α/E2F3 axis | [104] |
| LINC00518 | Enhances glycolysis by sponging miR-33a-3p, promoting HIF-1α/LDHA axis and increasing radioresistance | [105] |
| CCHE1 | Promotes glycolysis through FGFR1-LDHA complex, supporting melanoma progression and chemoresistance | [106] |
| circMYC (hsa_circ_0085533) | Increases glycolytic flux by sponging miR-1236, relieving LDHA repression | [107] |
| miR-33a | Acts as tumor suppressor by binding HIF-1α 3′UTR, inhibiting proliferation and glycolysis | [108,109] |
| SAMMSON | Impairs mitochondrial homeostasis, increases sensitivity to BRAF/MEK inhibitors by blocking metabolic adaptation | [110] |
| LENOX (LINC00518) | Promotes switch from glycolysis to OXPHOS following MAPK inhibition, contributing to drug resistance | [111] |
| Drugs | Target(s) | Mechanism of Action | Clinical Status | Cancer Type | Clinical Trial ID |
|---|---|---|---|---|---|
| Guadecitabine + Ipilimumab | DNMT1; CTLA-4 | Hypomethylating agent boosts tumor immunogenicity; CTLA-4 blockade activates T-cell response | Phase I completed | Metastatic melanoma | NCT02608437 Guadecitabine [149] |
| Decitabine + Vemurafenib | DNMT1; BRAFV600E | DNA demethylation delays MAPK-inhibitor resistance via epigenetic reprogramming | Phase Ib completed | BRAF-mutant melanoma | NCT01876641 [150] |
| Entinostat + Pembrolizumab | Class I HDAC; PD-1 | HDAC inhibition reduces MDSCs and increases antigen expression; potentiates PD-1 checkpoint blockade | Phase II (ENCORE-601) | Anti–PD-1–refractory melanoma | NCT02437136 [151] |
| CPI-1205 + Ipilimumab | EZH2; CTLA-4 | Reverses gene silencing via EZH2 inhibition; enhances tumor visibility and immune responsiveness | Phase I/II completed | Solid tumors incl. melanoma | NCT03525795 |
| Phenformin + Dabrafenib/Trametinib | Mitochondrial Complex I; BRAF/MEK | Biguanide blocks OXPHOS in resistant cells; MAPK inhibitors target glycolysis-reliant cells | Phase I completed (RP2D defined) | BRAFV600E melanoma | NCT03026517 [152] |
| Telaglenastat (CB-839) + Nivolumab | Glutaminase (GLS1); PD-1 | Glutamine metabolism inhibition reshapes redox and TME to increase T-cell effectiveness | Phase I/II completed | Advanced melanoma | NCT02771626 [153] |
| AZD3965 | MCT1 | Inhibits lactate export in glycolytic tumors | Phase I (suspended in melanoma) | Solid tumors and lymphomas | NCT01791595 [154] |
| INCB059872 + Epacadostat + Pembrolizumab | LSD1; IDO1; PD-1 | LSD1 inhibitor enhances tumor immunogenicity; IDO1 inhibition reduces immunosuppression; PD-1 blockade activates T-cell response | Phase I/II completed | Advanced solid tumors incl. melanoma | NCT02959437 [155] |
| Epacadostat + Pembrolizumab | IDO1; PD-1 | IDO1 inhibition to enhance PD-1 blockade efficacy in unselected melanoma patients | Phase III completed | Metastatic melanoma | NCT02752074 [156] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giuliani, S.; Accetta, C.; di Martino, S.; De Vitis, C.; Messina, E.; Pescarmona, E.; Fanciulli, M.; Ciliberto, G.; Mancini, R.; Falcone, I. Metabolic Reprogramming in Melanoma: An Epigenetic Point of View. Pharmaceuticals 2025, 18, 853. https://doi.org/10.3390/ph18060853
Giuliani S, Accetta C, di Martino S, De Vitis C, Messina E, Pescarmona E, Fanciulli M, Ciliberto G, Mancini R, Falcone I. Metabolic Reprogramming in Melanoma: An Epigenetic Point of View. Pharmaceuticals. 2025; 18(6):853. https://doi.org/10.3390/ph18060853
Chicago/Turabian StyleGiuliani, Stefano, Celeste Accetta, Simona di Martino, Claudia De Vitis, Elena Messina, Edoardo Pescarmona, Maurizio Fanciulli, Gennaro Ciliberto, Rita Mancini, and Italia Falcone. 2025. "Metabolic Reprogramming in Melanoma: An Epigenetic Point of View" Pharmaceuticals 18, no. 6: 853. https://doi.org/10.3390/ph18060853
APA StyleGiuliani, S., Accetta, C., di Martino, S., De Vitis, C., Messina, E., Pescarmona, E., Fanciulli, M., Ciliberto, G., Mancini, R., & Falcone, I. (2025). Metabolic Reprogramming in Melanoma: An Epigenetic Point of View. Pharmaceuticals, 18(6), 853. https://doi.org/10.3390/ph18060853