


Microorganisms 2026, 14(5), 1110; https://doi.org/10.3390/microorganisms14051110 - 13 May 2026
Abstract
Spinal infections in general, and infectious spondylodiscitis in particular, are increasingly diagnosed in the Western world, in recent decades. This rise in incidence is associated with an ageing population and with an increased availability of accurate diagnostic modalities. Even so, due to the
[...] Read more.
Spinal infections in general, and infectious spondylodiscitis in particular, are increasingly diagnosed in the Western world, in recent decades. This rise in incidence is associated with an ageing population and with an increased availability of accurate diagnostic modalities. Even so, due to the non-specific nature of clinical manifestations, and of the implicated blood and serum markers, there is a risk of underdiagnosis or misdiagnosis of the disease in its initial stages. Ionizing radiation methods, such as plain radiography (X-ray) and computed tomography (CT), are also not reliable in the early stages of the diseases, and the golden standard of imagistic diagnosis, magnetic resonance imaging (MRI), is not always available or requested. Still, MRI remains the most reliable method in most cases where there is a need for differential diagnosis with other pathologies, namely Andersson lesions, destructive spondyloarthropathy, erosive osteochondritis, micro-crystalline spondylitis, Modic 1 lesion, Charcot spinal arthropathy, osteoporotic fractures, SAPHO syndrome with spinal involvement, and Schmorl’s nodes. Infectious spondylodiscitis is caused by bacteria, and, less frequently, by fungi. Rare cases of parasitic causes have also been reported in the literature. Infectious spondylodiscitis of bacterial causes may be pyogenic, more frequently caused by Staphylococcus spp. or Streptococcus spp., or granulomatous, usually caused by Mycobacterium tuberculosis complex (MTBC) or from classical brucellosis. In all these cases, therapy may be conservative, with antibiotics, or surgical, when the former fails or in patients with significant spinal instability or other neurological manifestations. There are various surgical approaches, each with its own drawbacks, and usually used according to the preference of the attending physician. Even in cases of surgical treatment, antibiotic administration is prolonged, and it is important for a proper scheme to be selected based on antimicrobial susceptibility testing. However, given that in many cases, the causative agent cannot be identified, empirical treatment must be initiated. Finally, newer approaches, including the incorporation of antimicrobial substances, may offer better solutions for improving treatment and rehabilitation outcomes.
Full article
(This article belongs to the Special Issue The Epidemiology, Clinical Aspects, Treatment and Preventive Strategies for Infectious Diseases in the Era of Antimicrobial Resistance (AMR))
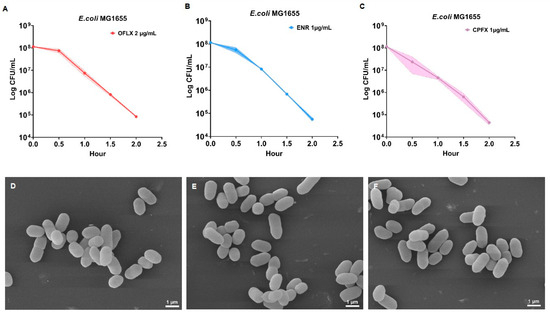
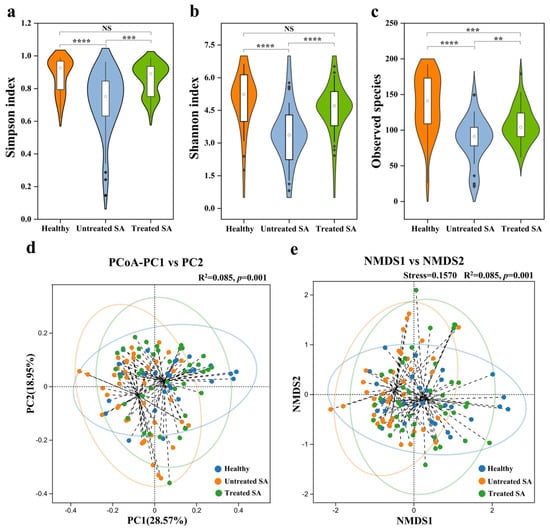
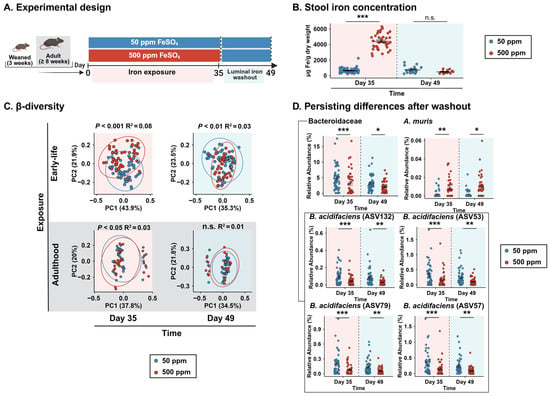
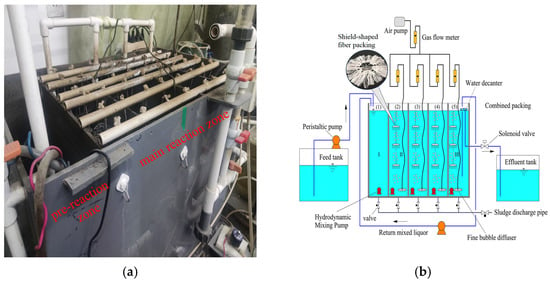

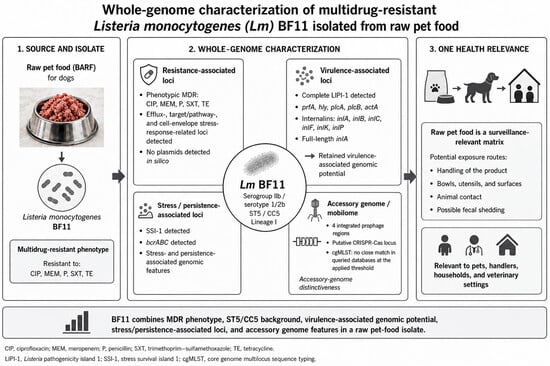
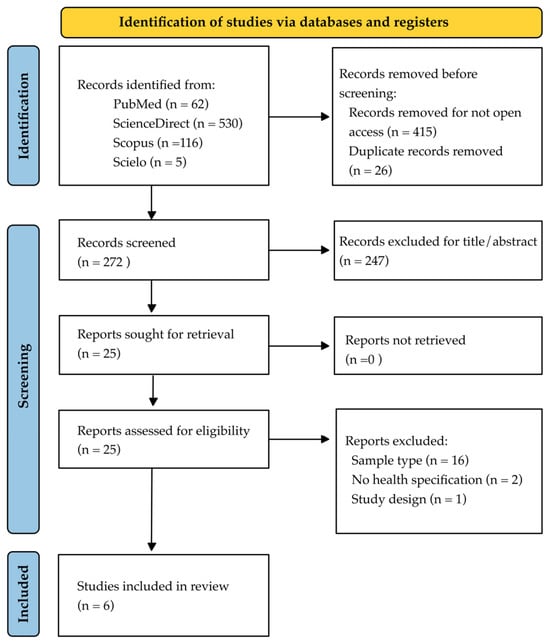

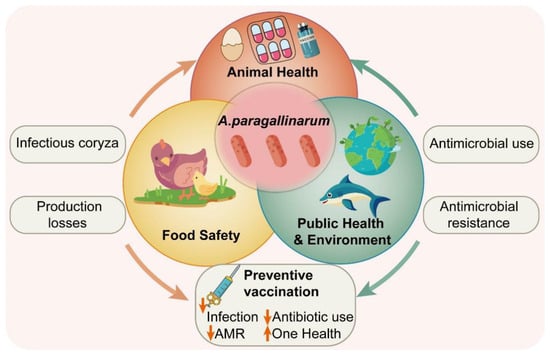






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}