



Animals 2026, 16(4), 644; https://doi.org/10.3390/ani16040644 - 17 Feb 2026
Viewed by 739
Abstract
►
Show Figures
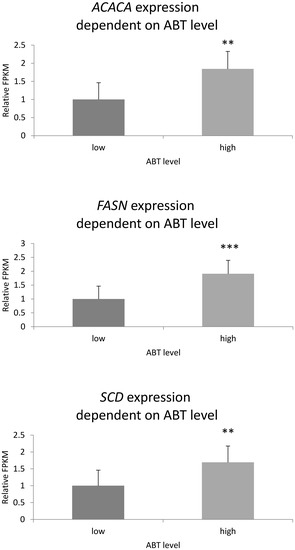
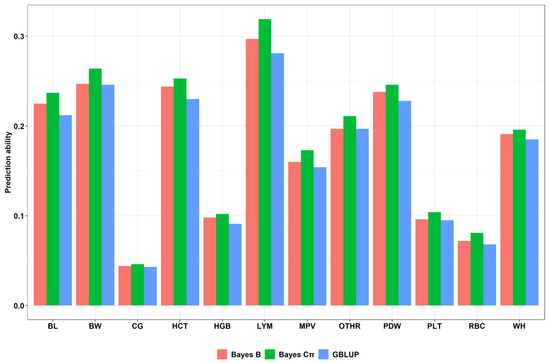
The economic performance of Holstein, Montbéliarde × Holstein, and Simmental × Holstein cows was evaluated using a bio-economic model. The results showed differences in income and costs between crossbred and pure Holstein groups. The Montbéliarde–Holstein crossbreds had the highest milk income ($2223) and
[...] Read more.
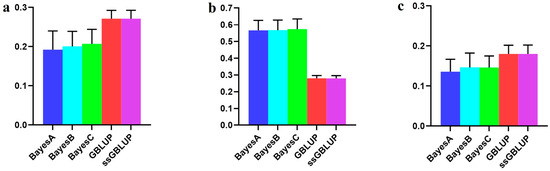
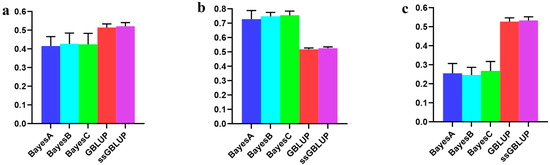
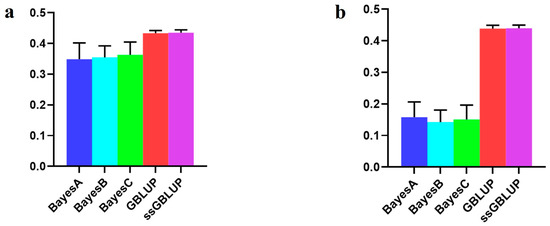
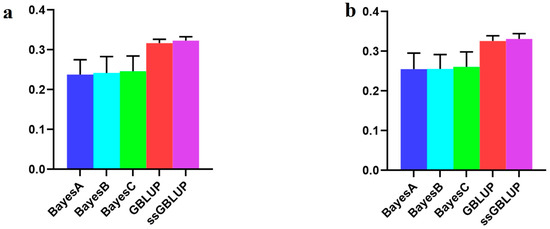
The economic performance of Holstein, Montbéliarde × Holstein, and Simmental × Holstein cows was evaluated using a bio-economic model. The results showed differences in income and costs between crossbred and pure Holstein groups. The Montbéliarde–Holstein crossbreds had the highest milk income ($2223) and profitability ($1002) compared to the Simmental crossbreds and pure Holstein. In contrast, pure Holstein made the most money from selling extra heifers, while both crossbred groups, especially Simmental × Holstein, made more money from selling male calves. Crossbred systems had much lower annual maintenance costs. Simmental × Holstein had variable costs that were 22% lower than those of pure Holstein. Trait-based economic evaluations demonstrated that Montbéliarde crossbreds exhibited the highest economic value for milk production, while Simmental crosses were more susceptible to prolonged calving intervals. Both crossbred groups had higher birth weights and growth rates than Holstein cows. Overall, these results provide strong evidence that crossbreeding Holstein with Montbéliarde maximizes overall profitability, while Simmental × Holstein also offers notable economic benefits through reduced maintenance costs and improved growth performance.
Full article
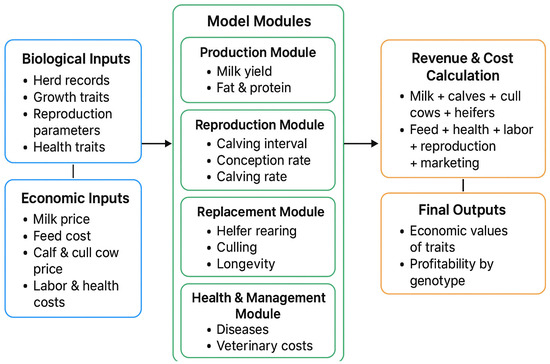
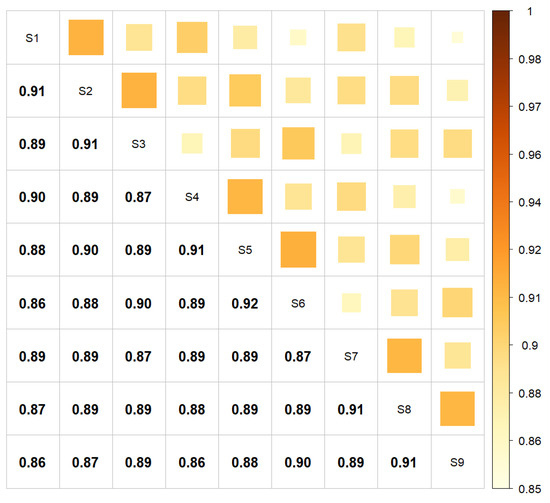
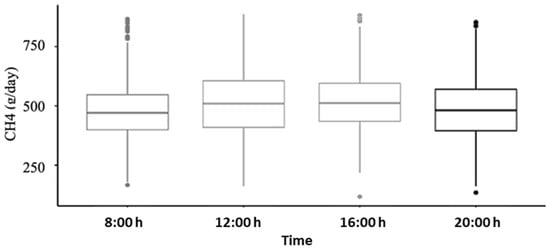
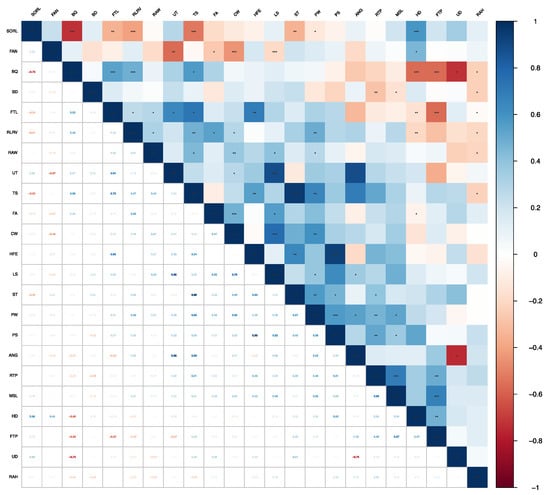
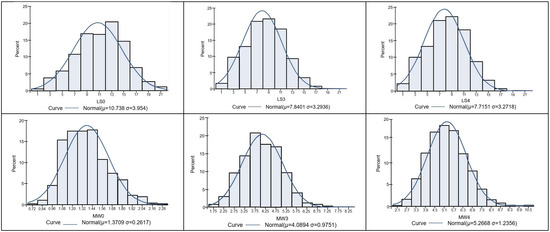
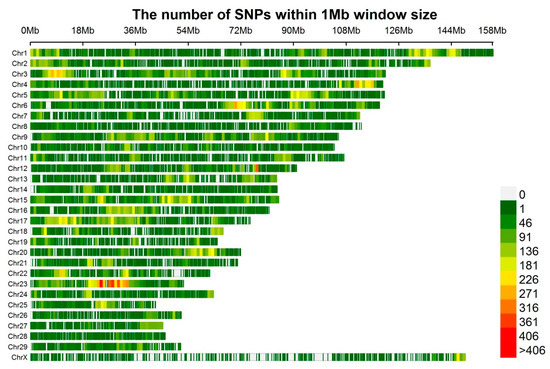
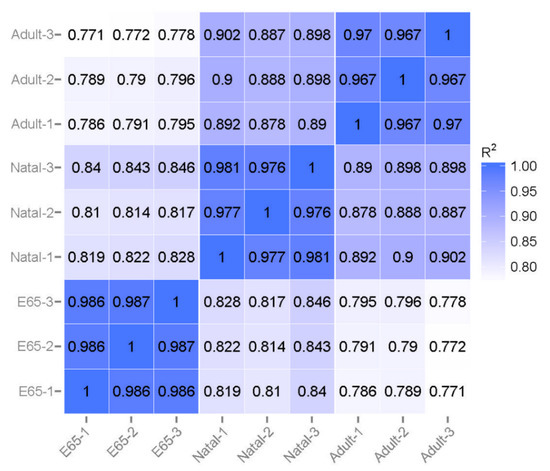
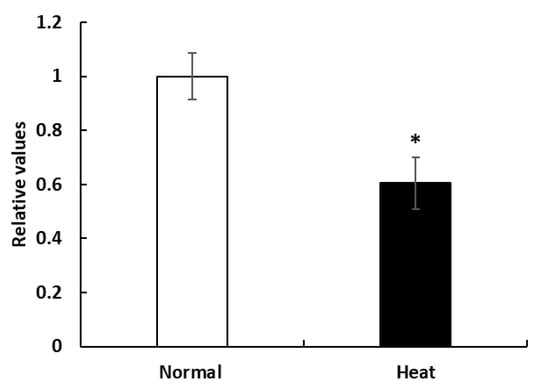
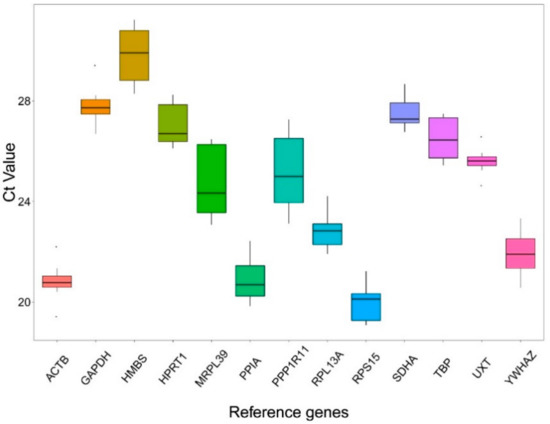
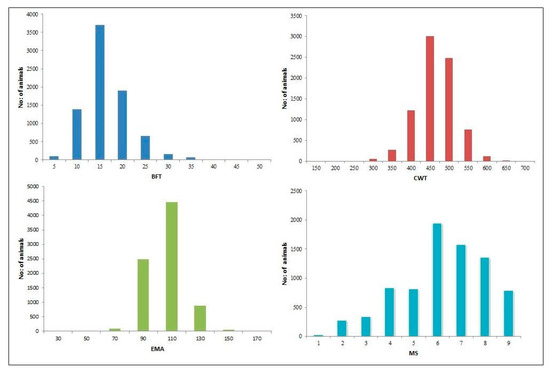
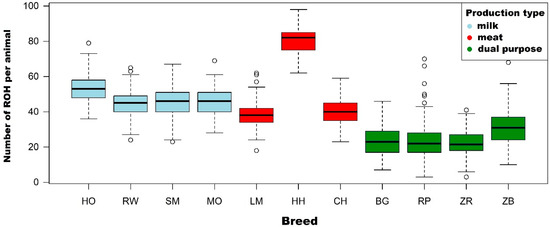
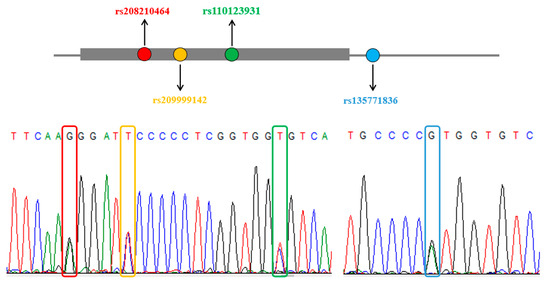
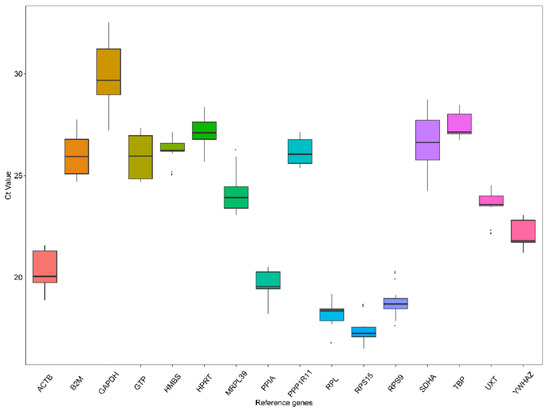
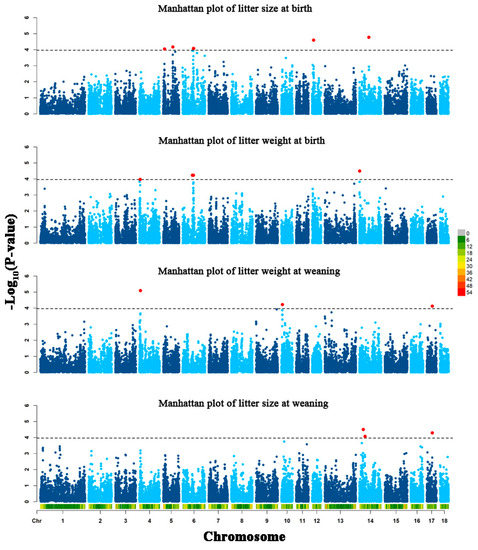
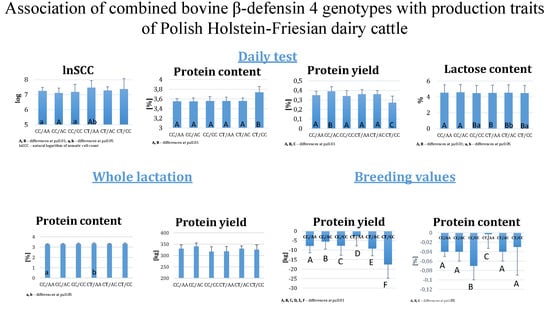
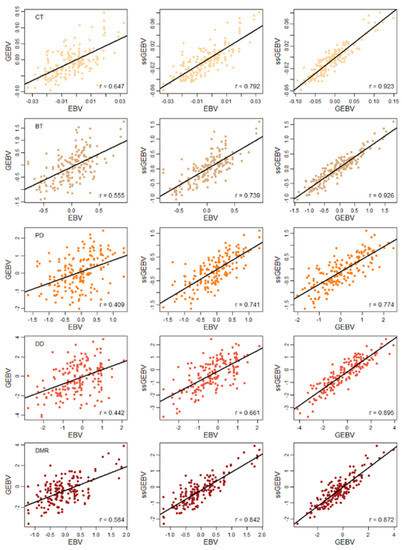
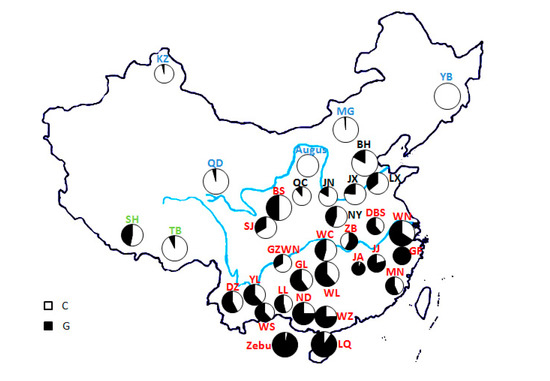
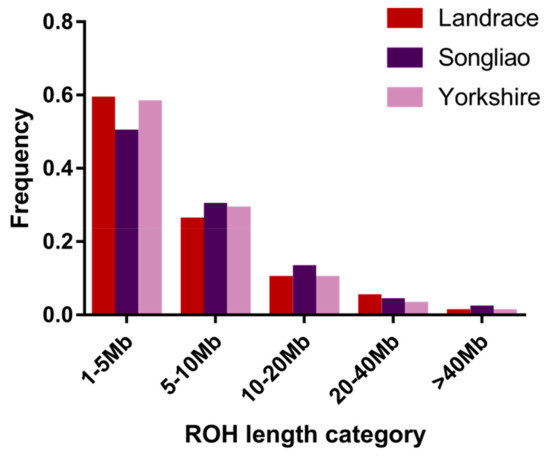
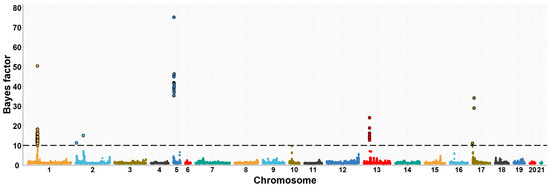
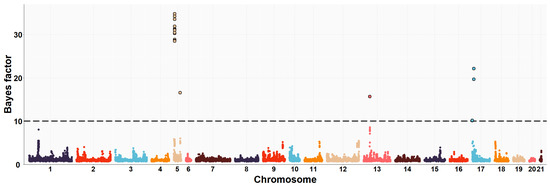
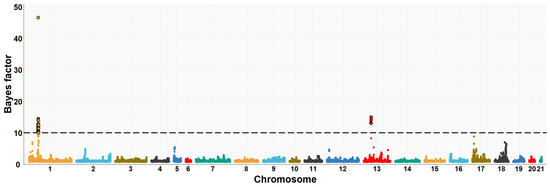
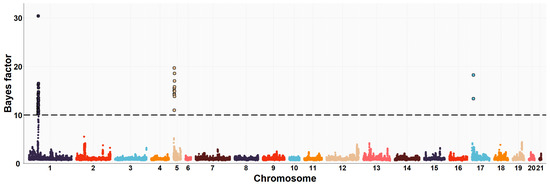
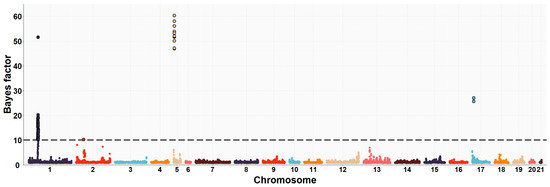
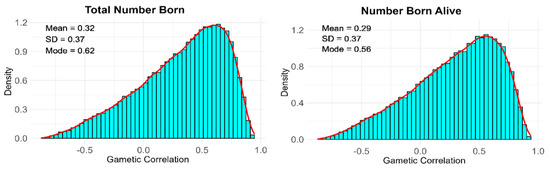
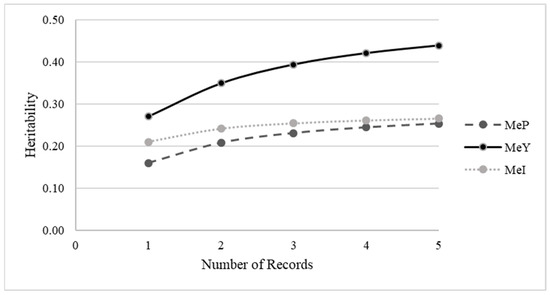
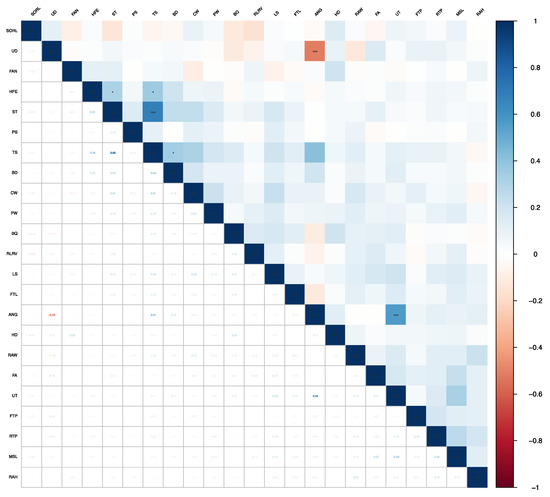
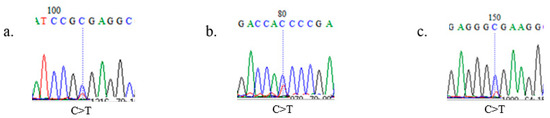
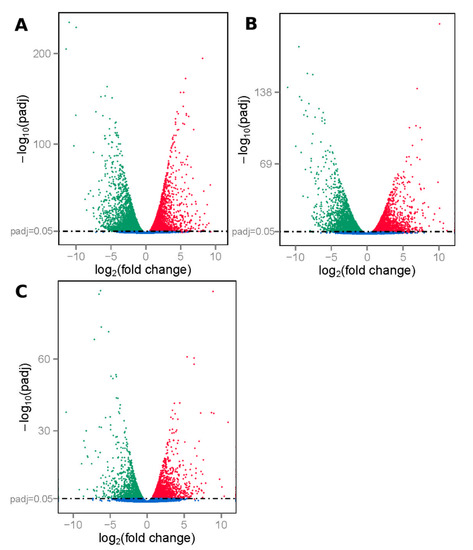
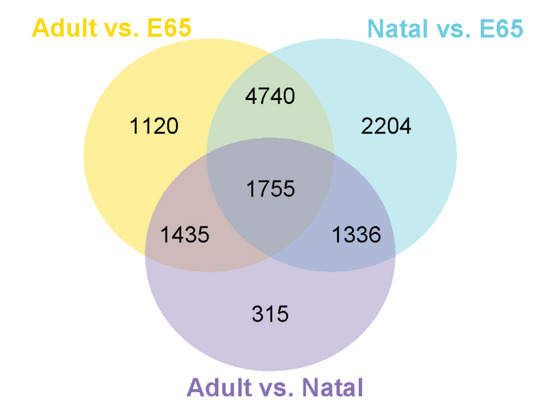
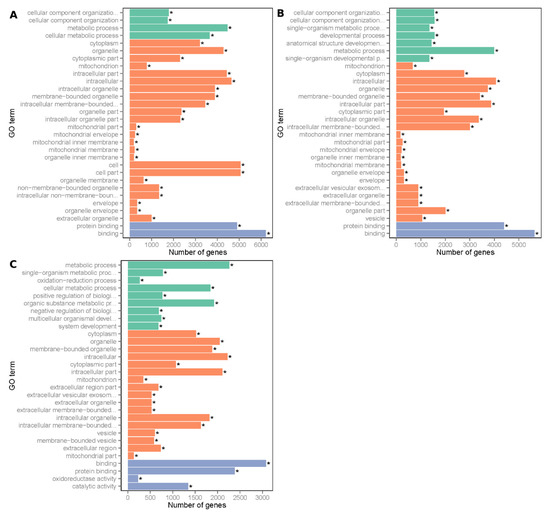
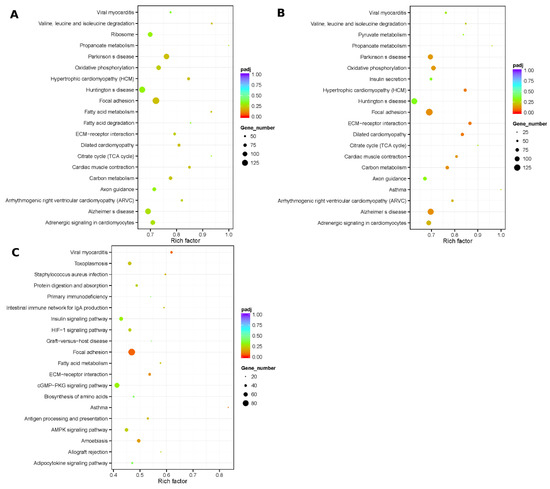
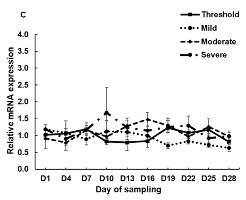
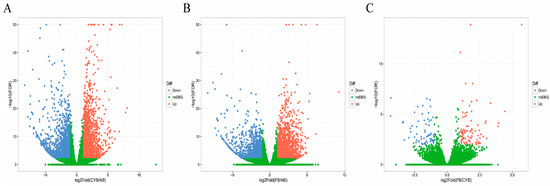
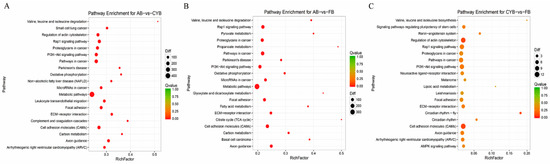
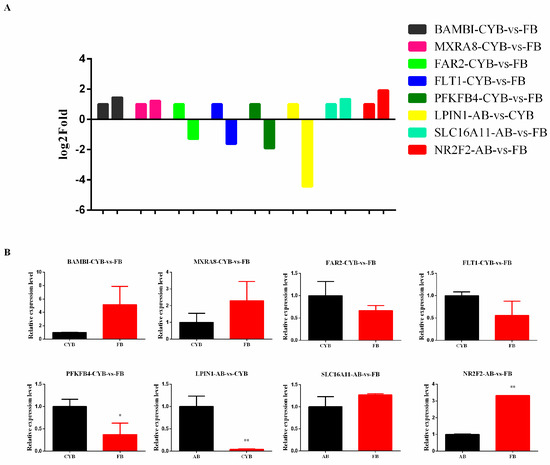
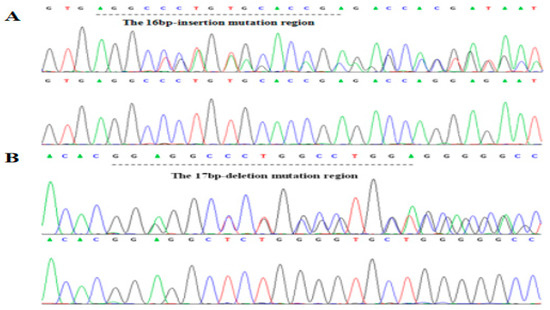
Figure 1





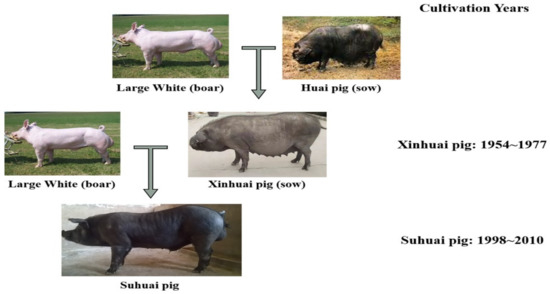
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}