
Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals, Phenotypes, and DNA Extraction
2.3. Whole-Genome Sequencing
2.4. Quality Control
2.5. Population Structure Analysis
2.6. Genome-Wide Association Studies
2.7. Functional Enrichment Analysis
3. Results
3.1. Phenotypic Data Analysis
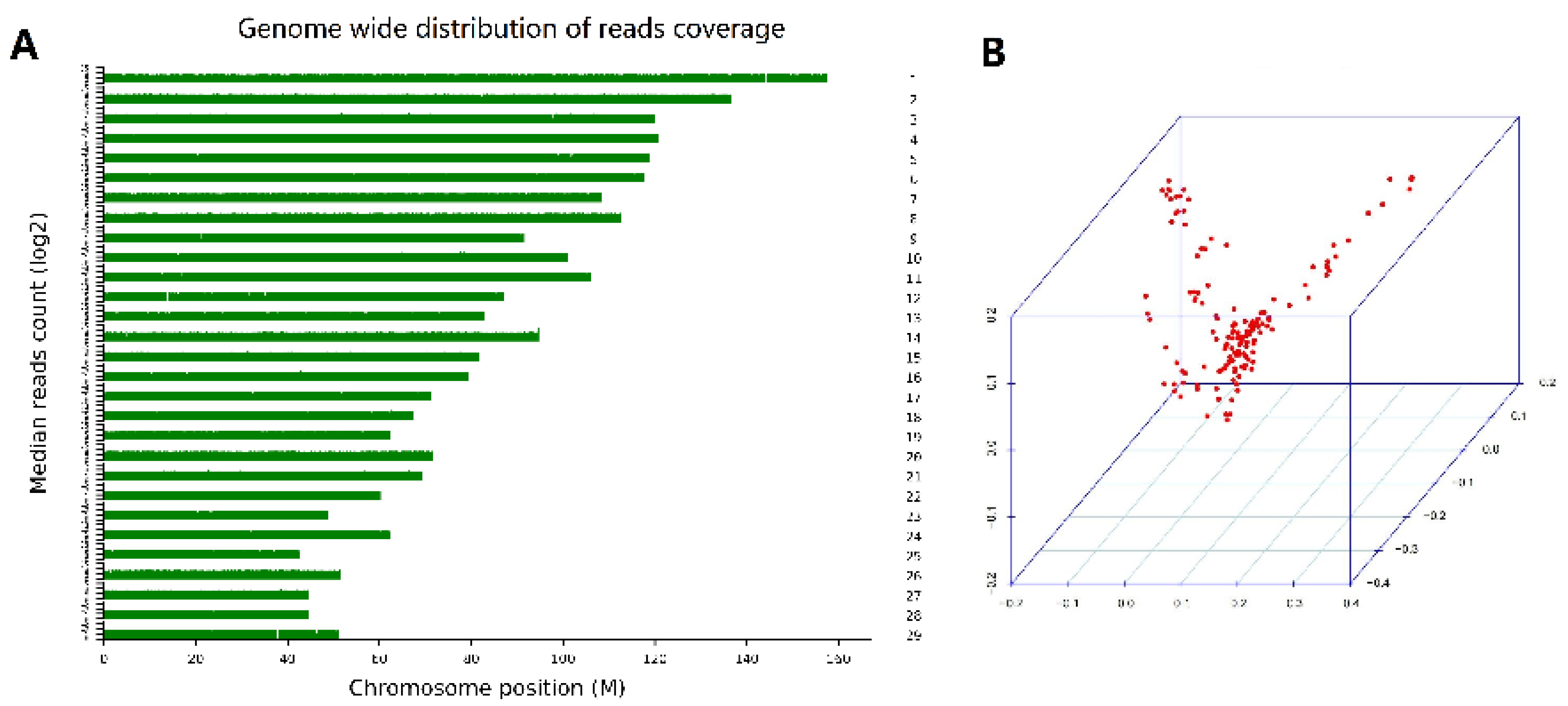
3.2. Genotypic Data
3.3. Population Structure Analysis
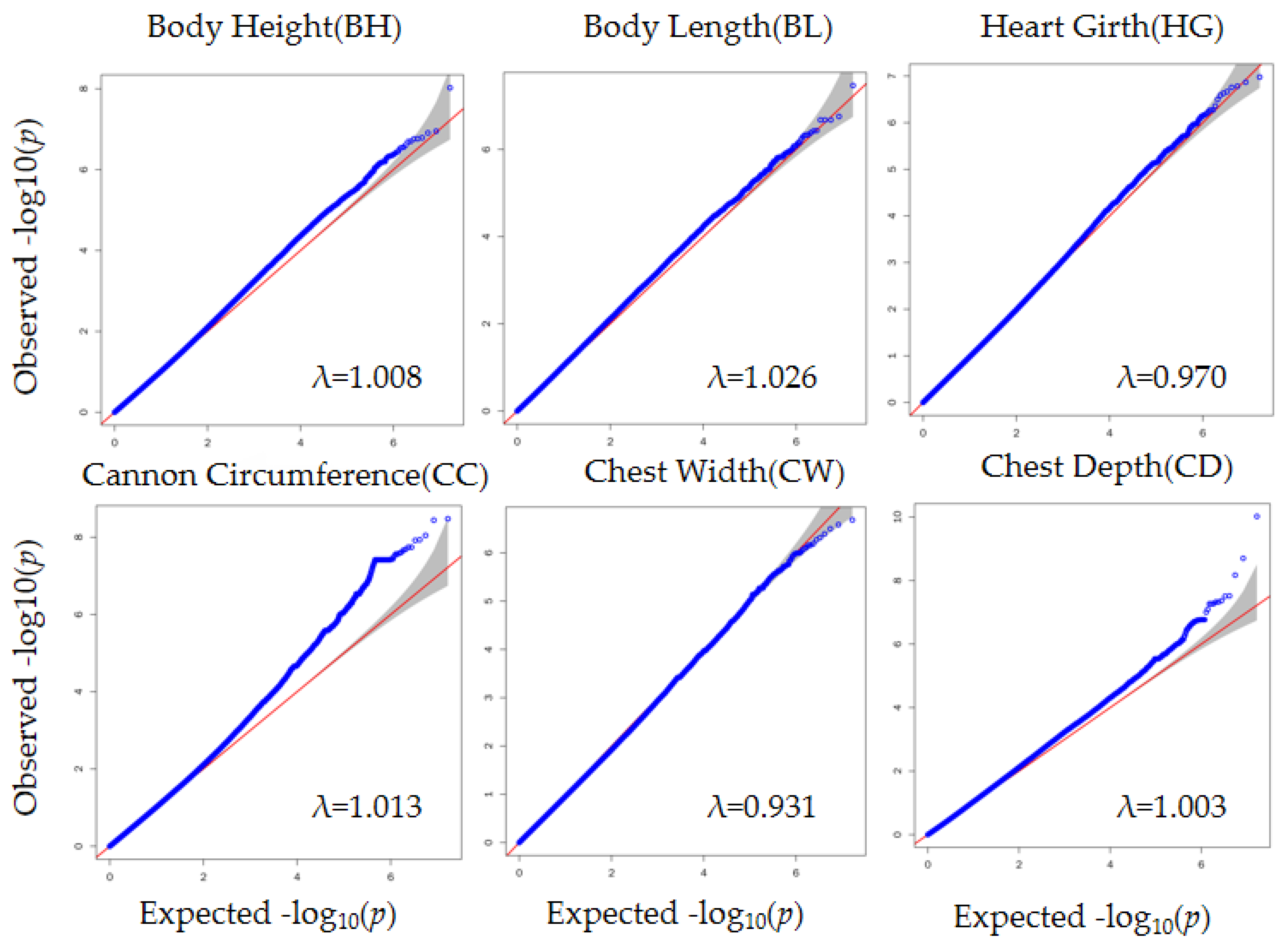
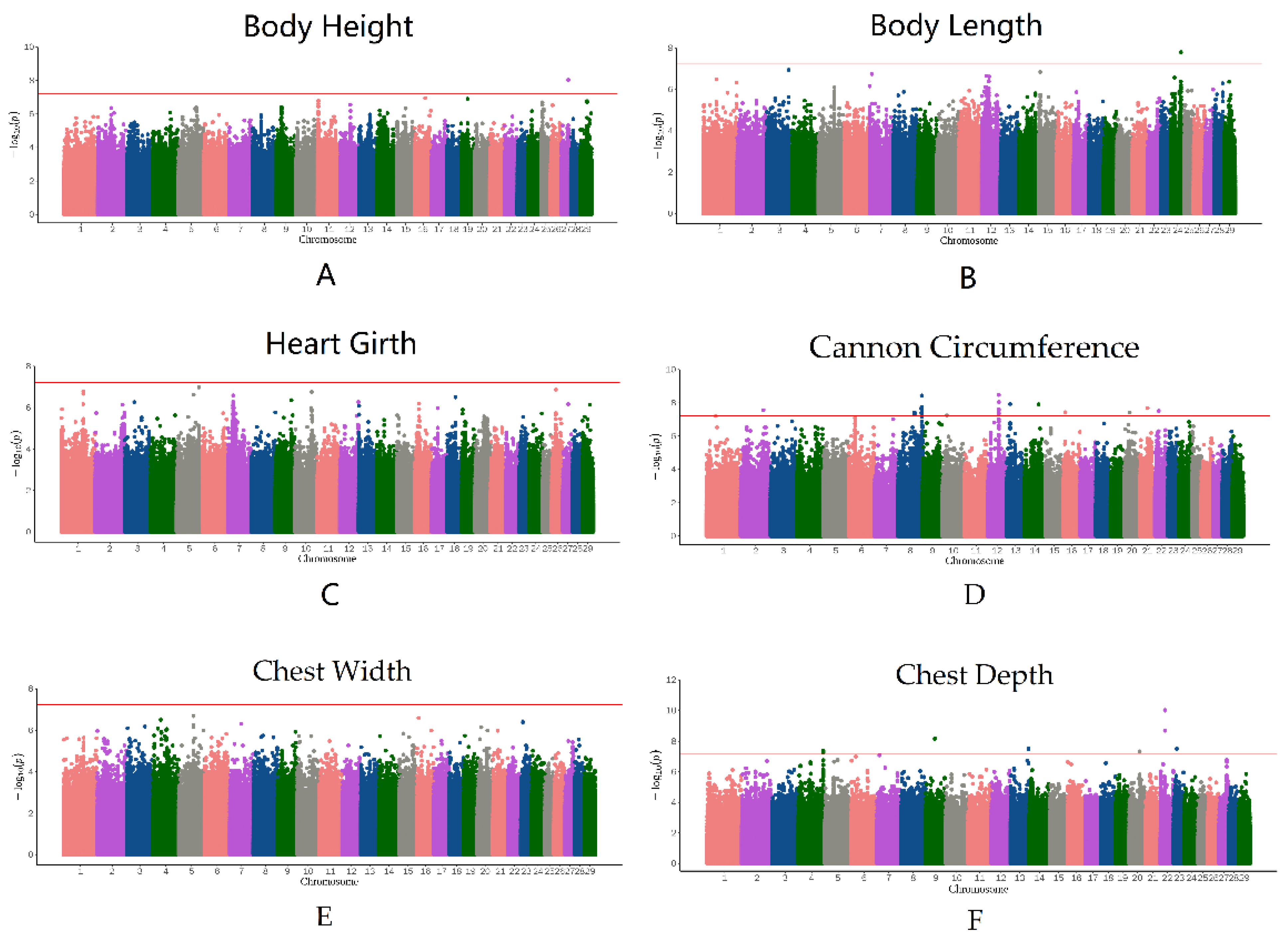
3.4. Genome-Wide Association Study
3.5. GO Enrichment and KEGG Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naderi, S.; Rezaei, H.R.; Pompanon, F.; Blum, M.G.B.; Negrini, R.; Naghash, H.R.; Balkiz, O.; Mashkour, M.; Gaggiotti, O.E.; Ajmone-Marsan, P.; et al. The goat domestication process inferred from large-scale mitochondrial DNA analysis of wild and domestic individuals. Proc. Natl. Acad. Sci. USA 2008, 105, 17659–17664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanda, J.S.; Taylor, D.G.; Murray, P.J.; Pegg, R.B.; Shand, P.J. Goat meat production: Present status and future possibilities. Asian Australas. J. Anim. Sci. 2003, 16, 1842–1852. [Google Scholar] [CrossRef]
- Pophiwa, P.; Webb, E.C.; Frylinck, L. A review of factors affecting goat meat quality and mitigating strategies. Small Rumin. Res. 2020, 183, 7. [Google Scholar] [CrossRef]
- Wei, C.H.; Lu, J.; Xu, L.Y.; Liu, G.; Wang, Z.G.; Zhao, F.P.; Zhang, L.; Han, X.; Du, L.X.; Liu, C.S. Genetic Structure of Chinese Indigenous Goats and the Special Geographical Structure in the Southwest China as a Geographic Barrier Driving the Fragmentation of a Large Population. PLoS ONE 2014, 9, 12. [Google Scholar] [CrossRef]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of selection signature provides a glimpse into important economic traits in goats (Capra hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef] [Green Version]
- Cam, M.A.; Olfaz, M.; Soydan, E. Body Measurements Reflect Body Weights and Carcass Yields in Karayaka Sheep. Asian J. Anim. Vet. Adv. 2010, 5, 120–127. [Google Scholar] [CrossRef]
- Yoshida, G.M.; Yanez, J.M. Multi-trait GWAS using imputed high-density genotypes from whole-genome sequencing identifies genes associated with body traits in Nile tilapia. BMC Genom. 2021, 22, 57. [Google Scholar] [CrossRef]
- Luigi-Sierra, M.G.; Landi, V.; Guan, D.; Delgado, J.V.; Castello, A.; Cabrera, B.; Marmol-Sanchez, E.; Alvarez, J.F.; Gomez-Carpio, M.; Martinez, A.; et al. A genome-wide association analysis for body, udder, and leg conformation traits recorded in Murciano-Granadina goats. J. Dairy Sci. 2020, 103, 11605–11617. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, C.; Guo, Y.; She, S.; Wang, B.; Jiang, Y.; Bai, Y.; Song, X.; Li, L.; Shi, L.; et al. Screening of Deletion Variants within the Goat PRDM6 Gene and Its Effects on Growth Traits. Animals 2020, 10, 208. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, L.; Wang, M.; Wang, K.; Li, J.; Yan, H.; Zhu, H.; Lan, X. Two indel variants of prolactin receptor (PRLR) gene are associated with growth traits in goat. Anim. Biotechnol. 2020, 31, 314–323. [Google Scholar] [CrossRef]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.Y.; Li, J.; Zhu, H.J.; Liu, J.W.; Dong, S.W.; Li, L.P.; Qu, L.; Chen, H.; Song, X.Y.; Lan, X.Y. Deletion mutation within the goat PPP3CA gene identified by GWAS significantly affects litter size. Reprod. Fertil. Dev. 2021, 33, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Liu, X.X.; Gebreselassie, G.R.M.; Abied, A.D.; Ma, Q.; Ma, Y.H. Genome-wide association analysis reveals the genetic locus for high reproduction trait in Chinese Arbas Cashmere goat. Genes Genom. 2020, 42, 893–899. [Google Scholar] [CrossRef]
- Guo, J.Z.; Jiang, R.; Mao, A.; Liu, G.E.; Zhan, S.Y.; Li, L.; Zhong, T.; Wang, L.J.; Cao, J.X.; Chen, Y.; et al. Genome-wide association study reveals 14 new SNPs and confirms two structural variants highly associated with the horned/polled phenotype in goats. BMC Genom. 2021, 22, 10. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.M.; Palhiere, I.; Ricard, A.; Tosser-Klopp, G.; Rupp, R. Genome Wide Association Study Identifies New Loci Associated with Undesired Coat Color Phenotypes in Saanen Goats. PLoS ONE 2016, 11, 15. [Google Scholar] [CrossRef] [PubMed]
- Tilahun, Y.; Gipson, T.A.; Alexander, T.; McCallum, M.L.; Hoyt, P.R. Genome-Wide Association Study towards Genomic Predictive Power for High Production and Quality of Milk in American Alpine Goats. Int. J. Genom. 2020, 2020, 12. [Google Scholar] [CrossRef]
- Scholtens, M.; Jiang, A.; Smith, A.; Littlejohn, M.; Lehnert, K.; Snell, R.; Lopez-Villalobos, N.; Garrick, D.; Blair, H. Genome-wide association studies of lactation yields of milk, fat, protein and somatic cell score in New Zealand dairy goats. J. Anim. Sci. Biotechnol. 2020, 11, 55. [Google Scholar] [CrossRef]
- Wang, K.; Liu, X.; Qi, T.; Hui, Y.; Yan, H.; Qu, L.; Lan, X.; Pan, C. Whole-genome sequencing to identify candidate genes for litter size and to uncover the variant function in goats (Capra hircus). Genomics 2021, 113 Pt 1, 142–150. [Google Scholar] [CrossRef]
- Wu, P.X.; Wang, K.; Zhou, J.; Chen, D.J.; Yang, Q.; Yang, X.D.; Liu, Y.H.; Feng, B.; Jiang, A.N.; Shen, L.Y.; et al. GWAS on Imputed Whole-Genome Resequencing From Genotyping-by-Sequencing Data for Farrowing Interval of Different Parities in Pigs. Front. Genet. 2019, 10, 1012. [Google Scholar] [CrossRef]
- Sanchez, M.P.; Guatteo, R.; Davergne, A.; Saout, J.; Grohs, C.; Deloche, M.C.; Taussat, S.; Fritz, S.; Boussaha, M.; Blanquefort, P.; et al. Identification of the ABCC4, IER3, and CBFA2T2 candidate genes for resistance to paratuberculosis from sequence-based GWAS in Holstein and Normande dairy cattle. Genet. Sel. Evol. 2020, 52, 17. [Google Scholar] [CrossRef] [Green Version]
- Tribout, T.; Croiseau, P.; Lefebvre, R.; Barbat, A.; Boussaha, M.; Fritz, S.; Boichard, D.; Hoze, C.; Sanchez, M.P. Confirmed effects of candidate variants for milk production, udder health, and udder morphology in dairy cattle. Genet. Sel. Evol. 2020, 52, 1–26. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, S.; Vandenplas, J.; van Eeuwijk, F.A.; Bouwman, A.C.; Lopes, M.S.; Veerkamp, R.F. Imputation to whole-genome sequence using multiple pig populations and its use in genome-wide association studies. Genet. Sel. Evol. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talouarn, E.; Bardou, P.; Palhiere, I.; Oget, C.; Clement, V.; VarGoats, C.; Tosser-Klopp, G.; Rupp, R.; Robert-Granie, C. Genome wide association analysis on semen volume and milk yield using different strategies of imputation to whole genome sequence in French dairy goats. BMC Genet. 2020, 21, 19. [Google Scholar] [CrossRef] [Green Version]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T.; et al. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, S.; Josse, J.; Husson, F. FactoMineR: An R package for multivariate analysis. J. Stat. Softw. 2008, 25, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Browning, B.L.; Zhou, Y.; Browning, S.R. A One-Penny Imputed Genome from Next-Generation Reference Panels. Am. J. Hum. Genet. 2018, 103, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh Bi, I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhiere, I.; Crisa, A.; Catillo, G.; Steri, R.; et al. Signatures of selection and environmental adaptation across the goat genome post-domestication. Genet. Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef] [PubMed]
- Colli, L.; Milanesi, M.; Talenti, A.; Bertolini, F.; Chen, M.H.; Crisa, A.; Daly, K.G.; Del Corvo, M.; Guldbrandtsen, B.; Lenstra, J.A.; et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 2018, 50, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kijas, J.W.; Ortiz, J.S.; McCulloch, R.; James, A.; Brice, B.; Swain, B.; Tosser-Klopp, G.; the International Goat Genome Consortium. Genetic diversity and investigation of polledness in divergent goat populations using 52088 SNPs. Anim. Genet. 2013, 44, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.Q.; Du, X.Y.; Zhang, W.G.; Tosser-Klopp, G.; Wang, J.H.; Yang, S.; Liang, J.; et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; He, X.Y.; Jiang, Y.T.; Lan, R.; Li, M.; Li, Z.M.; Yang, W.F.; Hong, Q.H.; Chu, M.X. Combined approaches to reveal genes associated with litter size in Yunshang black goats. Anim. Genet. 2020, 51, 924–934. [Google Scholar] [CrossRef]
- Yang, B.G.; Yuan, Y.; Zhou, D.K.; Ma, Y.H.; Mahrous, K.F.; Wang, S.Z.; He, Y.M.; Duan, X.H.; Zhang, W.Y.; E, G. Genome-wide selection signal analysis of Australian Boer goat reveals artificial selection imprinting on candidate genes related to muscle development. Anim. Genet. 2021, 52, 550–555. [Google Scholar] [CrossRef]
- Li, X.K.; Su, R.; Wan, W.T.; Zhang, W.G.; Jiang, H.Z.; Qiao, X.; Fan, Y.X.; Zhang, Y.J.; Wang, R.J.; Liu, Z.H.; et al. Identification of selection signals by large-scale whole-genome resequencing of cashmere goats. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Visscher, P.M.; Wray, N.R.; Zhang, Q.; Sklar, P.; McCarthy, M.I.; Brown, M.A.; Yang, J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017, 101, 5–22. [Google Scholar] [CrossRef] [Green Version]
- E, G.X.; Duan, X.H.; Zhang, J.H.; Huang, Y.F.; Zhao, Y.J.; Na, R.S.; Zhao, Z.Q.; Ma, Y.H.; Chu, M.X.; Basang, W.D.; et al. Genome-wide selection signatures analysis of litter size in Dazu black goats using single-nucleotide polymorphism. 3 Biotech 2019, 9, 336. [Google Scholar] [CrossRef]
- E, G.-X.; Zhu, Y.B.; Basang, W.D.; Na, R.S.; Han, Y.G.; Zeng, Y. Comparative and selection sweep analysis of CNV was associated to litter size in Dazu black goats. Anim. Biotechnol. 2021, 32, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Gross, J.M.; Calabrese, C.; Iwakura, Y.; Lamkanfi, M.; Vogel, P.; Kanneganti, T.D. Critical role for inflammasome-independent IL-1beta production in osteomyelitis. Proc. Natl. Acad. Sci. USA 2014, 111, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Cai, X.; Yu, H.; Xu, Q.; Li, X.; Yang, Y.; Meng, X.; Huang, C.; Li, J. PSTPIP2 attenuates joint damage and suppresses inflammation in adjuvant-induced arthritis. Eur. J. Pharmacol. 2019, 859, 172558. [Google Scholar] [CrossRef] [PubMed]
- Sztacho, M.; Segeletz, S.; Sanchez-Fernandez, M.A.; Czupalla, C.; Niehage, C.; Hoflack, B. BAR Proteins PSTPIP1/2 Regulate Podosome Dynamics and the Resorption Activity of Osteoclasts. PLoS ONE 2016, 11, e0164829. [Google Scholar] [CrossRef] [PubMed]
- Chio, A.; Schymick, J.C.; Restagno, G.; Scholz, S.W.; Lombardo, F.; Lai, S.L.; Mora, G.; Fung, H.C.; Britton, A.; Arepalli, S.; et al. A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.J.; Bruckner, R.J.; Paulo, J.A.; Kazak, L.; Long, J.Z.; Mina, A.I.; Deng, Z.; LeClair, K.B.; Hall, J.A.; Hong, S.; et al. Cdkal1, a type 2 diabetes susceptibility gene, regulates mitochondrial function in adipose tissue. Mol. Metab. 2017, 6, 1212–1225. [Google Scholar] [CrossRef]
- Takesue, Y.; Wei, F.Y.; Fukuda, H.; Tanoue, Y.; Yamamoto, T.; Chujo, T.; Shinojima, N.; Yano, S.; Morioka, M.; Mukasa, A.; et al. Regulation of growth hormone biosynthesis by Cdk5 regulatory subunit associated protein 1-like 1 (CDKAL1) in pituitary adenomas. Endocr. J. 2019, 66, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lv, L.; Zheng, N.; Li, R.; Yang, R.; Li, T.; Li, Y.; Liu, Y.; Luo, H.; Li, X.; et al. Suppression of Sox4 protects against myocardial ischemic injury by reduction of cardiac apoptosis in mice. J. Cell. Physiol. 2021, 236, 1094–1104. [Google Scholar] [CrossRef]
- Qin, Y.; He, L.D.; Sheng, Z.J.; Yong, M.M.; Sheng, Y.S.; Dong, X.W.; Wen, T.W.; Ming, Z.Y. Increased CCL19 and CCL21 levels promote fibroblast ossification in ankylosing spondylitis hip ligament tissue. BMC Musculoskelet. Disord. 2014, 15, 316. [Google Scholar] [CrossRef] [Green Version]
- Nan, H.; Ichinose, Y.; Tanaka, M.; Koh, K.; Ishiura, H.; Mitsui, J.; Mizukami, H.; Morimoto, M.; Hamada, S.; Ohtsuka, T.; et al. UBAP1 mutations cause juvenile-onset hereditary spastic paraplegias (SPG80) and impair UBAP1 targeting to endosomes. J. Hum. Genet. 2019, 64, 1055–1065. [Google Scholar] [CrossRef]
- Wang, J.; Hou, Y.; Qi, L.; Zhai, S.; Zheng, L.; Han, L.; Guo, Y.; Zhang, B.; Miao, P.; Lou, Y.; et al. Autosomal dominant hereditary spastic paraplegia caused by mutation of UBAP1. Neurogenetics 2020, 21, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yao, X.; Yang, H.; Deng, K.; Guo, Y.; Zhang, T.; Zhang, G.; Wang, F. Role of FGF9 in sheep testis steroidogenesis during sexual maturation. Anim. Reprod. Sci. 2018, 197, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Roth, T.; Abbott, M.; Ho, L.; Wattanachanya, L.; Nissenson, R.A. Osteoblast-derived FGF9 regulates skeletal homeostasis. Bone 2017, 98, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demonbreun, A.R.; Wyatt, E.J.; Fallon, K.S.; Oosterbaan, C.C.; Page, P.G.; Hadhazy, M.; Quattrocelli, M.; Barefield, D.Y.; McNally, E.M. A gene-edited mouse model of limb-girdle muscular dystrophy 2C for testing exon skipping. Dis. Model. Mech. 2019, 13, dmm040832. [Google Scholar] [CrossRef] [Green Version]
- Cox, G.A.; Mahaffey, C.L.; Nystuen, A.; Letts, V.A.; Frankel, W.N. The mouse fidgetin gene defines a new role for AAA family proteins in mammalian development. Nat. Genet. 2000, 26, 198–202. [Google Scholar] [CrossRef]
- Ge, X.; Li, Z.; Zhou, Z.; Xia, Y.; Bian, M.; Yu, J. Circular RNA SIPA1L1 promotes osteogenesis via regulating the miR-617/Smad3 axis in dental pulp stem cells. Stem. Cell Res. Ther. 2020, 11, 364. [Google Scholar] [CrossRef]


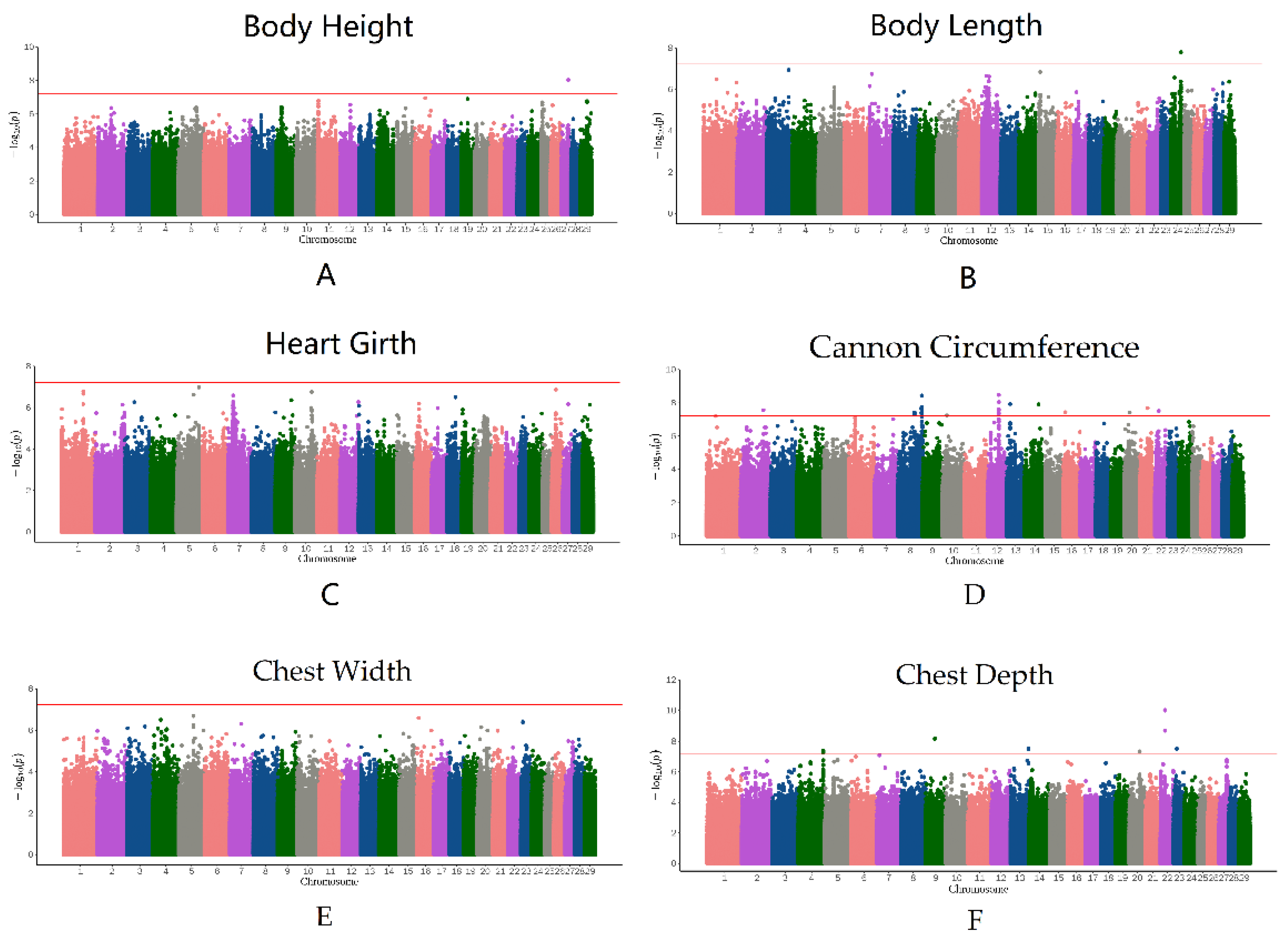

{kind=link}
{kind=link}
{kind=link}
| Trait | Max (cm) | Min (cm) | Mean (cm) | Var | Std. Dev | CV |
|---|---|---|---|---|---|---|
| BH | 75.5 | 50.4 | 63.52 | 12.70 | 3.56 | 5.6% |
| BL | 78.5 | 54.3 | 67.27 | 79.16 | 4.38 | 6.5% |
| CC | 10.3 | 6.8 | 8.13 | 0.33 | 0.58 | 7.0% |
| CD | 38.3 | 20.5 | 29.78 | 6.06 | 2.46 | 8.2% |
| CW | 23.9 | 8.2 | 17.13 | 4.68 | 2.16 | 12.0% |
| HG | 99.5 | 68.7 | 81.65 | 29.60 | 5.44 | 6.0% |
| Goat (n) | Raw Bases (Gb) 1 | Reads Mapped to the Reference 1 | Total Reads 1 | Mapping Ratio (%) 1 | Coverage Depth 1 |
|---|---|---|---|---|---|
| 150 | 17.83 | 116 293 649 | 117 573 225 | 99.06 | 6.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, B.; Sun, R.; Fang, X.; Zhang, J.; Zhao, Z.; Huang, D.; Zhao, Y.; Zhao, Y. Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats. Animals 2022, 12, 548. https://doi.org/10.3390/ani12050548
Gu B, Sun R, Fang X, Zhang J, Zhao Z, Huang D, Zhao Y, Zhao Y. Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats. Animals. 2022; 12(5):548. https://doi.org/10.3390/ani12050548
Chicago/Turabian StyleGu, Bowen, Ruifan Sun, Xingqiang Fang, Jipan Zhang, Zhongquan Zhao, Deli Huang, Yuanping Zhao, and Yongju Zhao. 2022. "Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats" Animals 12, no. 5: 548. https://doi.org/10.3390/ani12050548
APA StyleGu, B., Sun, R., Fang, X., Zhang, J., Zhao, Z., Huang, D., Zhao, Y., & Zhao, Y. (2022). Genome-Wide Association Study of Body Conformation Traits by Whole Genome Sequencing in Dazu Black Goats. Animals, 12(5), 548. https://doi.org/10.3390/ani12050548