Genome-Wide Linkage Disequilibrium and the Extent of Effective Population Sizes in Six Chinese Goat Populations Using a 50K Single Nucleotide Polymorphism Panel


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
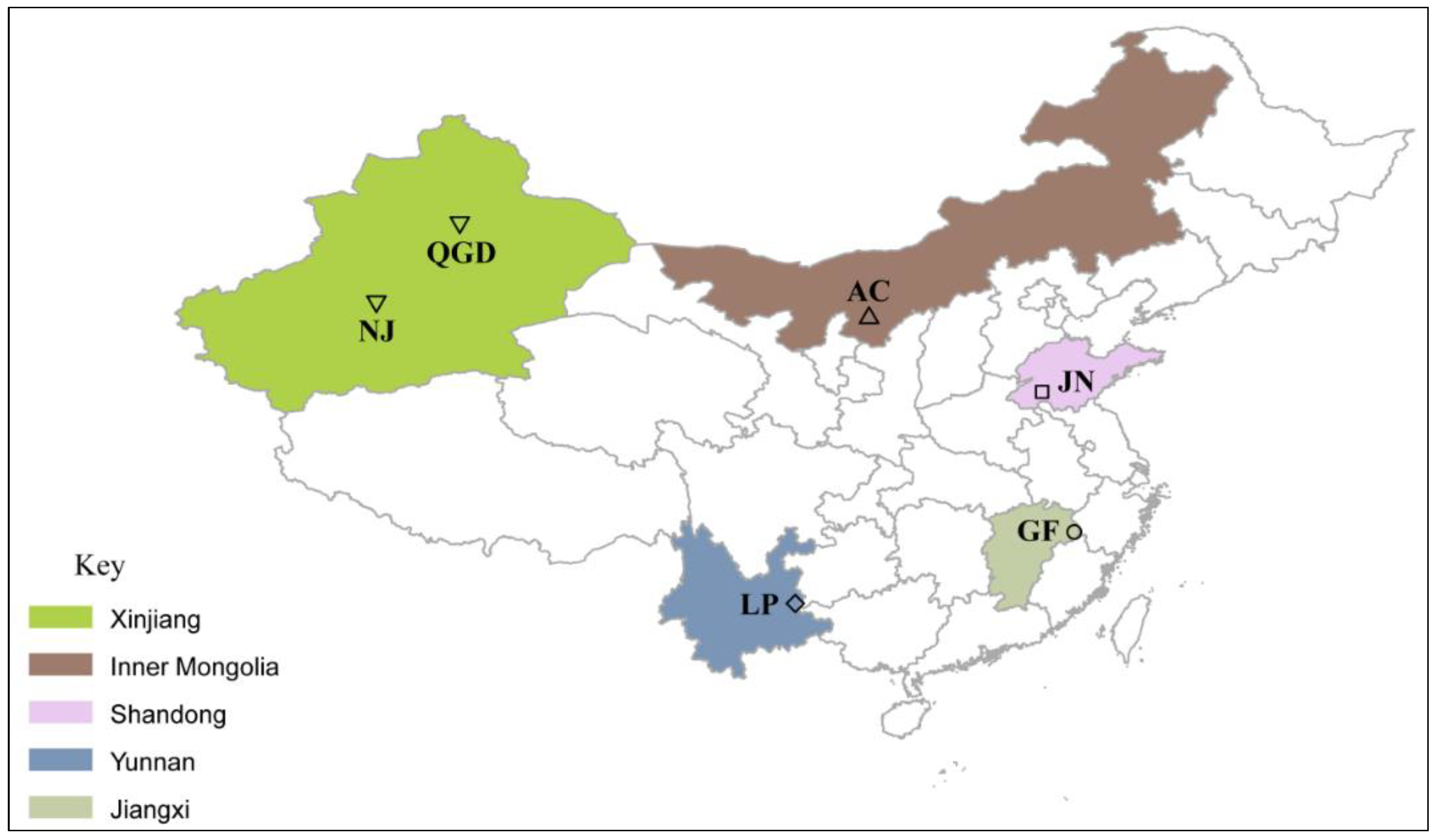
2.1. Animals and Sampling Units
2.2. Genotyping and Quality Control
2.3. Population Analyses
2.4. Minor Allele Frequncy and Inbreeding Coefficient
2.5. Linkage Disequilibrium (LD) Analysis
2.6. Effective Population Size (Ne)
3. Results
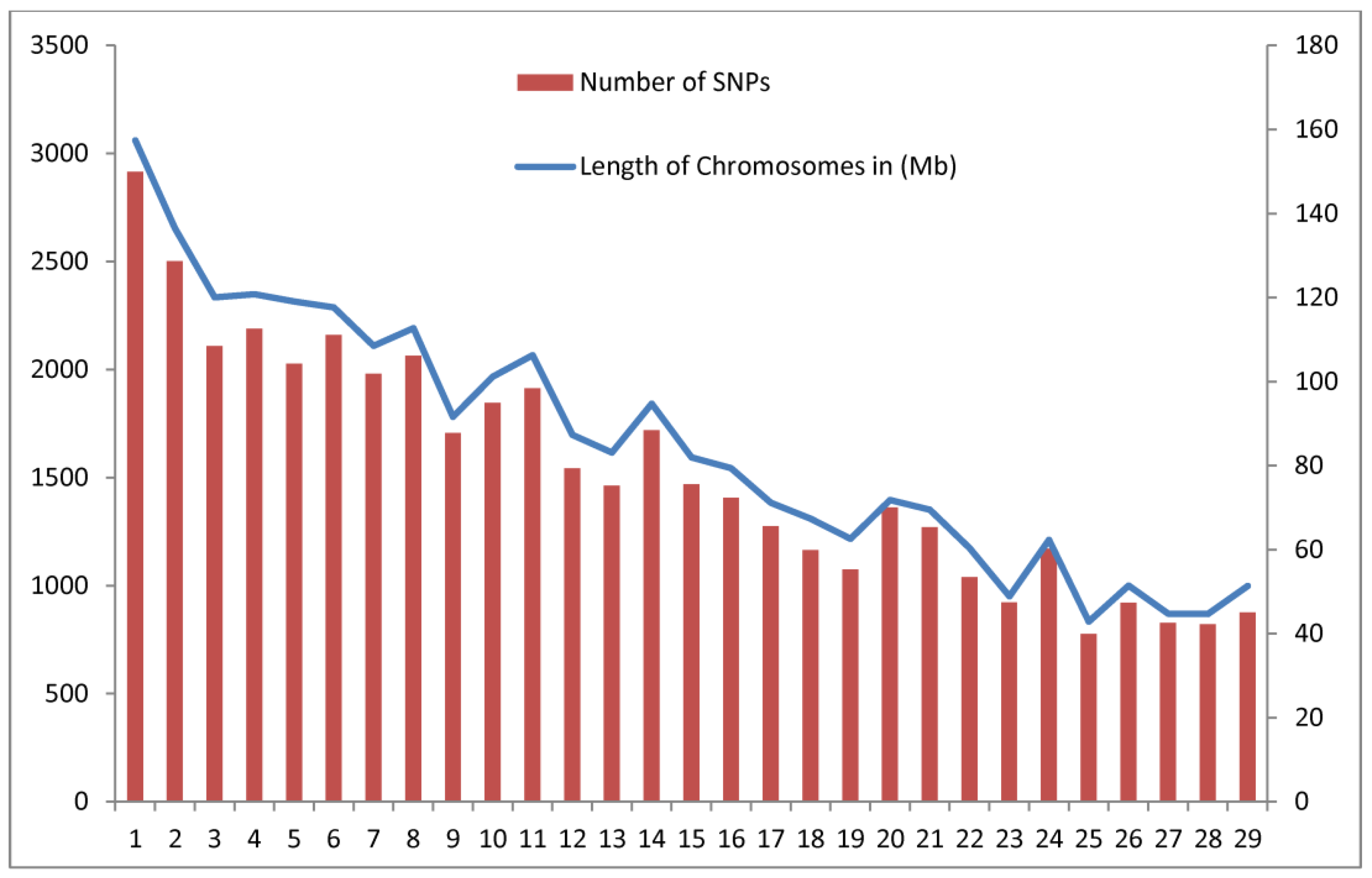
3.1. Descriptive Statistics
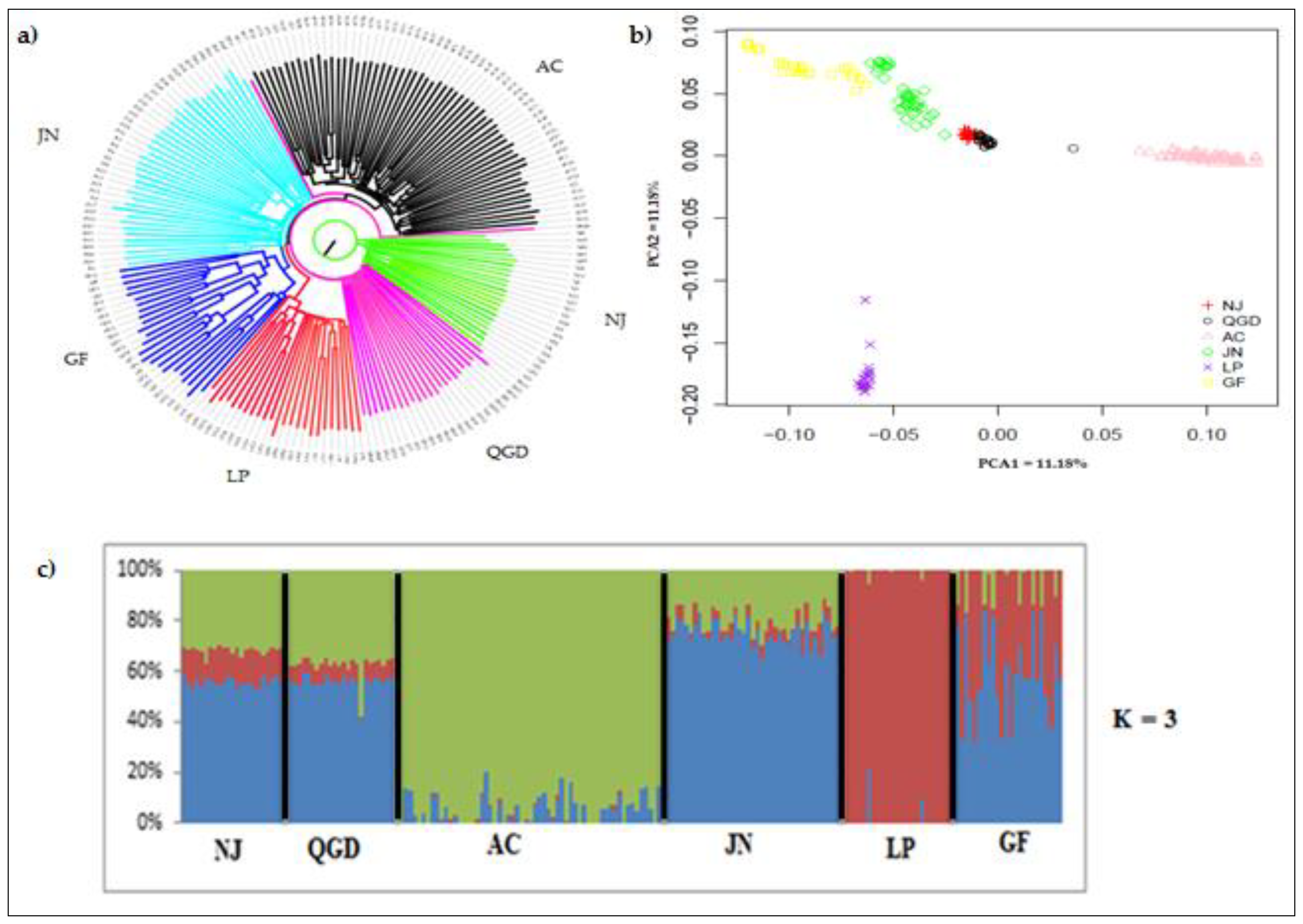
3.2. Population Genetic Structure and Admixture
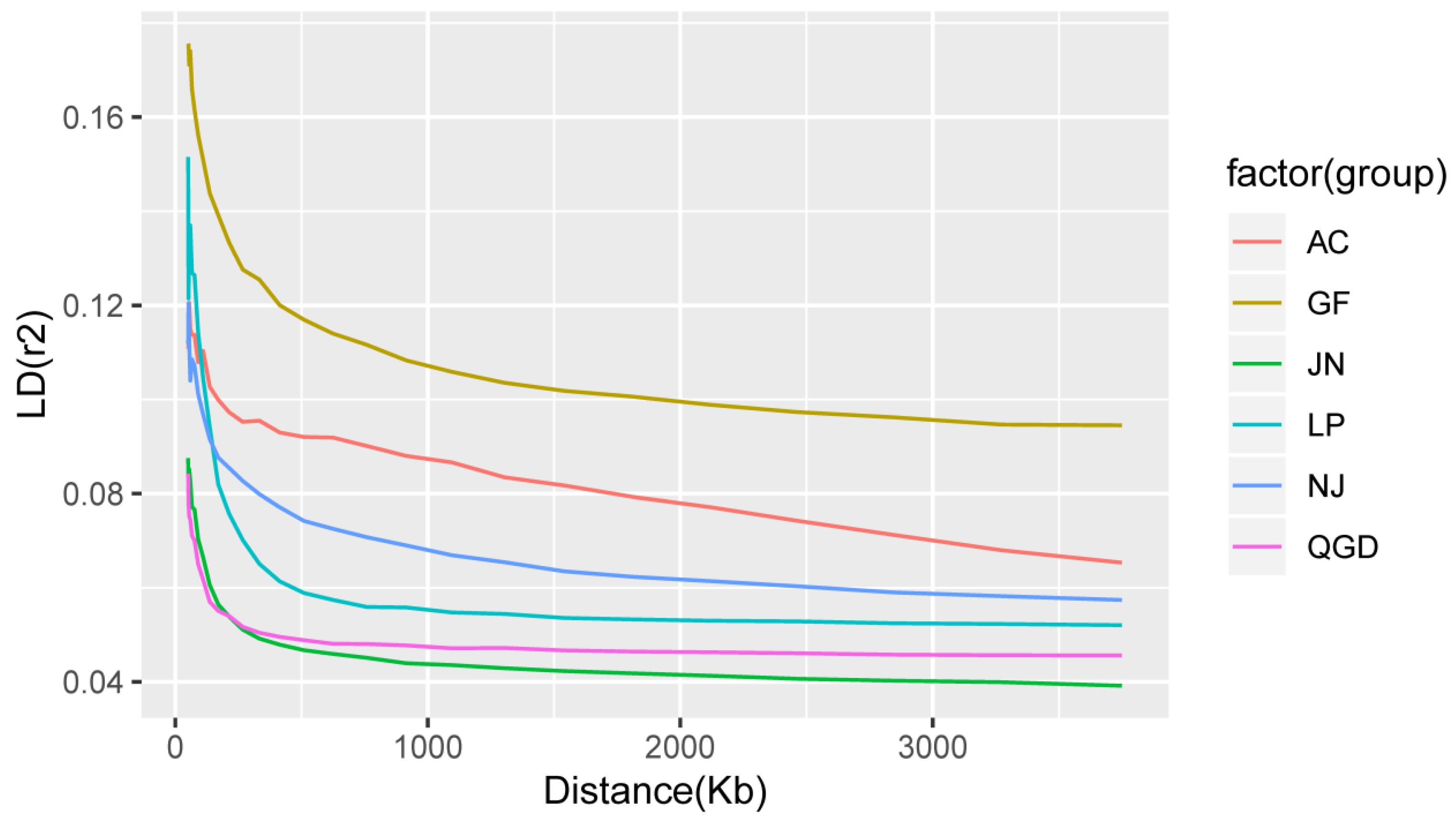
3.3. Minor Allele Frequency, Linkage Disequilibrium, and Inbreeding Coefficient
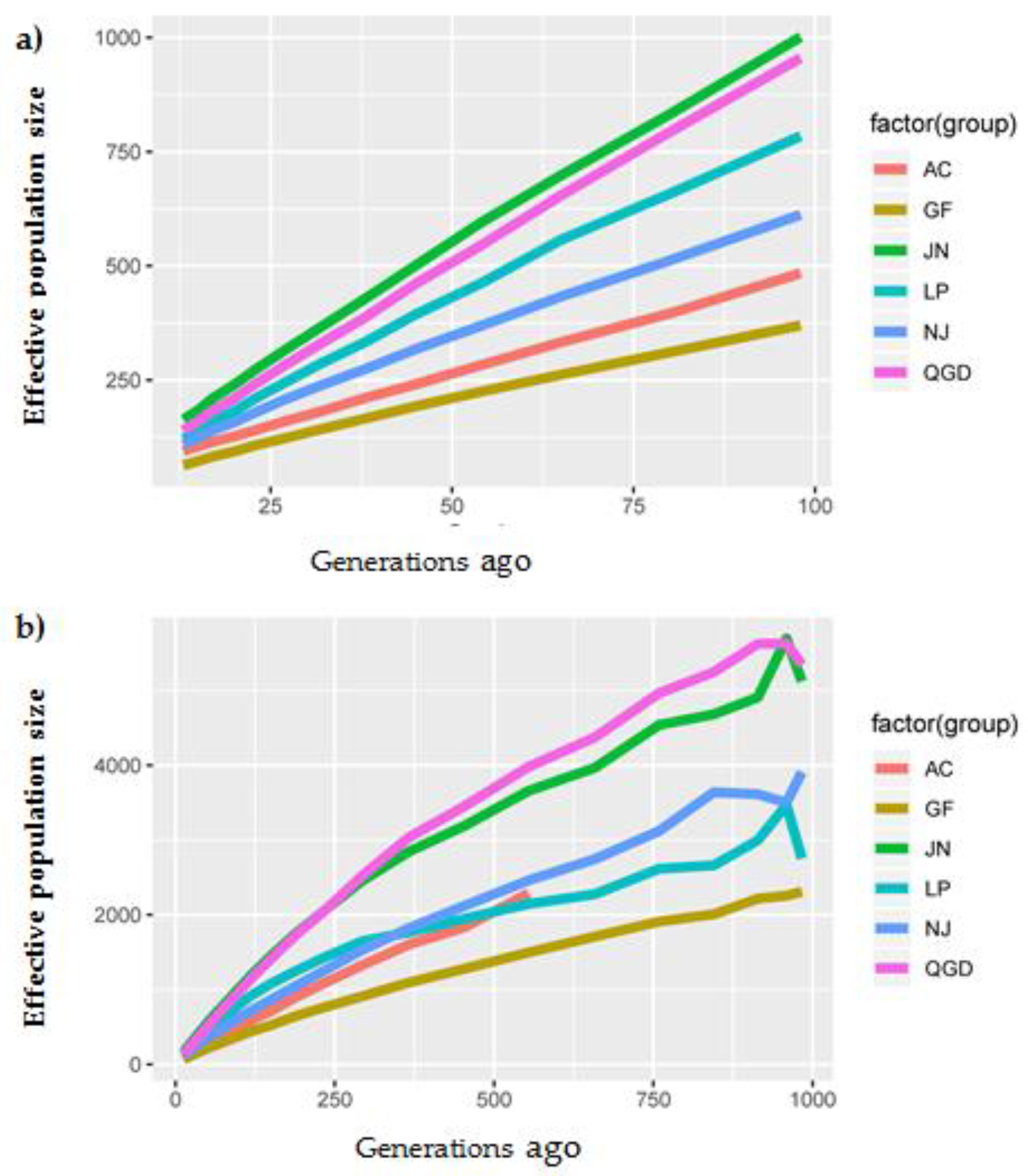
3.4. Effective Population Size (Ne)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Data Accessibility
References
- MacHugh, D.E.; Bradley, D.G. Livestock genetic origins: Goats buck the trend. Proc. Natl. Acad. Sci. USA 2001, 98, 5382–5384. [Google Scholar] [CrossRef] [PubMed]
- Colli, L.; Milanesi, M.; Talenti, A.; Bertolini, F.; Chen, M.; Crisà, A.; Daly, K.G.; Corvo, M.D.; Guldbrandtsen, B.; Lenstra, J.A.; et al. Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes. Genet. Sel. Evol. 2018, 50, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M. Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 2009, 136, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Park, L. Linkage Disequilibrium Decay and Past Population History in the Human Genome. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Sharma, A.; Lee, J.S.; Dang, C.G.; Sudrajad, P.; Kim, H.C.; Yeon, S.H.; Kang, H.S.; Hong, S.K. Genome Wide Association Studies in Livestock—A Review. Asian-Australas. J. Anim. Sci. 2015, 28, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; He, S.; Chen, L.; Li, W.; Di, J.; Liu, M. Estimates of linkage disequilibrium and effective population sizes in Chinese Merino (Xinjiang type) sheep by genome-wide SNPs. Gen. Genom. 2017, 39, 733–745. [Google Scholar] [CrossRef] [PubMed]
- Al-Mamun, H.A.; Clark, S.A.; Kwan, P.; Gondro, C. Genome-wide linkage disequilibrium and genetic diversity in five populations of Australian domestic sheep. Genet. Sel. Evolut. 2015, 47, 90. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, G.; Zeng, T.; Wei, C.; Zhang, L.; Wang, H.; Zhang, S.; Liu, R.; Liu, Z.; Du, L. Estimations of genomic linkage disequilibrium and effective population sizes in three sheep populations. Livest. Sci. 2014, 170, 22–29. [Google Scholar] [CrossRef]
- Deng, T.; Liang, A.; Liu, J.; Hua, G.; Ye, T.; Liu, S. Genome-Wide SNP Data Revealed the Extent of Linkage Disequilibrium, Persistence of Phase and Effective Population Size in Purebred and Crossbred Buffalo Populations. Front. Genet. 2019, 9, 688. [Google Scholar] [CrossRef]
- Nicoloso, L.; Bomba, L.; Colli, L.; Negrini, R.; Milanesi, M.; Mazza, R.; Sechi, T.; Frattini, S.; Talenti, A.; Coizet, B.; et al. Genetic diversity of Italian goat breeds assessed with a medium-density SNP chip. Genet. Sel. Evolut. 2015, 47, 62. [Google Scholar] [CrossRef]
- Alvarenga, A.B.; Rovadoscki, G.A.; Petrini, J.; Coutinho, L.L.; Morota, G.; Spangler, M.L.; Pinto, L.F.B.; Carvalho, G.G.P.; Mourão, G.B. Linkage disequilibrium in Brazilian Santa Inês breed, Ovis aries. Sci. Rep. 2018, 8, 8851. [Google Scholar] [CrossRef] [PubMed]
- Shina, D.; Cho, K.; Park, K.D.; Lee, H.J.; Kim, H. Accurate estimation of effective population size in the Korean dairy cattle based on linkage disequilibrium corrected by genomic relationship matrix. Asian-Australas. J. Anim. Sci. 2013, 26, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drogemuller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Mdladla, K.; Dzomba, E.F.; Huson, H.J.; Muchadeyi, F.C. Population genomic structure and linkage disequilibrium analysis of South African goat breeds using genome-wide SNP data. Anim. Genet. 2016, 47, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Uimari, P.; Tapio, M. Extent of linkage disequilibrium and effective population size in Finnish Landrace and Finnish Yorkshire pig breeds. J. Anim. Sci. 2011, 89, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Muse, S.V. PowerMarker : An integrated analysis environment for genetic marker analysis. Bioinformatics 2005, 21, 2128–2129. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Lewontin, R.C. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 1964, 49, 49–67. [Google Scholar]
- VanLiere, J.M.; Rosenberg, N.A. Mathematical properties of the r2 measure of linkage disequilibrium. Theor. Popul. Biol. 2008, 74, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Sved, J.A. Linkage Disequilibrium of Chromosome Segments. Theor. Popul. Biol. 1971, 141, 125–141. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hayes, B.J.; Visscher, P.M.; McPartlan, H.C.; Goddard, M.E. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Berihulay, H.; Li, Y.; Liu, X.; Gebreselassie, G.; Islam, R.; Liu, W.; Jiang, L.; Ma, Y. Genetic diversity and population structure in multiple Chinese goat populations using a panel. Anim. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lipkin, E.; Straus, K.; Stein, R.T.; Bagnato, A.; Schiavini, F.; Fontanesi, L.; Russo, V.; Medugorac, I.; Foerster, M.; Sölkner, J.; et al. Extensive Long-Range and Nonsyntenic Linkage Disequilibrium in Livestock Populations: Deconstruction of a Conundrum. Genetics 2009, 181, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Zapata, C. The D’ measure of overall gametic disequilibrium between pairs of multiallelic loci. Evolution 2000, 54, 1809–1812. [Google Scholar]
- Wray, N.R. Allele frequencies and the r2 measure of linkage disequilibrium: Impact on design and interpretation of association studies. Twin Res. Hum. Genet. 2005, 8, 87–94. [Google Scholar] [CrossRef]
- Khatkar, M.S.; Nicholas, F.W.; Collins, A.R.; Zenger, K.R.; Cavanagh, J.A.L.; Barris, W.; Schnabel, R.D.; Taylor, J.F.; Raadsma, H.W. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genom. 2008, 9, 187. [Google Scholar] [CrossRef]
- Getinet, M. Molecular Characterization of Ethiopian Indigenous Goat Populations: Genetic Diversity and Structure, Demographic Dynamics and Assessment of the Kisspeptin Gene Polymorphism. Ph.D. Thesis, Addis Ababa University, Addis Ababa, Ethiopia, 2016. [Google Scholar]
- Mustafa, H.; Ahmad, N.; Heather, H.J.; Eui-soo, K.; Khan, W.A.; Ajmal, A.; Javed, K.; Pasha, T.N.; Ali, A.; Kim, J.J.; et al. Whole genome study of linkage disequilibrium in Sahiwal cattle. S. Afr. J. Anim. Sci. 2018, 48, 353–360. [Google Scholar] [CrossRef]
- Makina, S.O.; Taylor, J.F.; Marle-Köster, E.; Muchadeyi, F.C.; Makgahlela, M.L.; MacNeil, M.D.; Maiwashe, A. Extent of Linkage Disequilibrium and Effective Population Size in Four South African Sanga Cattle Breeds. Front. Genet. 2015, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Townsend, S.M.; Jamieson, I.G. Molecular and pedigree measures of relatedness provide similar estimates of inbreeding depression in a bottlenecked population. J. Evolut. Biol. 2013, 26, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Reich, D.E.; Reich, D.E.; Cargill, M.; Cargill, M.; Bolk, S.; Bolk, S.; Ireland, J.; Ireland, J.; Sabeti, P.C.; Sabeti, P.C.; et al. Linkage disequilibrium in the human genome. Nature 2001, 411, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Meadows, J.R.S.; Chan, E.K.F.; Kijas, J.W. Linkage disequilibrium compared between five populations of domestic sheep. BMC Genet. 2008, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Visser, C.; Lashmar, S.F.; Marle-köster, E.V.; Poli, M.A.; Allain, D. Genetic Diversity and Population Structure in South African, French and Argentinian Angora Goats from Genome-Wide SNP Data. PLoS ONE 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lashmar, S.F.; Visser, C. SNP-based genetic diversity of South African commercial dairy and fiber goat breeds. Small Rumin. Res. 2016, 136, 65–71. [Google Scholar] [CrossRef]
- Brito, L.F.; Kijas, J.W.; Ventura, R.V.; Sargolzaei, M.; Porto-Neto, L.R.; Cánovas, A.; Feng, Z.; Jafarikia, M.; Schenkel, F.S. Genetic diversity and signatures of selection in various goat breeds revealed by genome-wide SNP markers. BMC Genom. 2017, 18, 229. [Google Scholar] [CrossRef] [PubMed]
- McKay, S.D.; Schnabel, R.D.; Murdoch, B.M.; Matukumalli, L.K.; Aerts, J.; Coppieters, W.; Crews, D.; Neto, E.; Gill, C.A.; Gao, C.; et al. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 2007, 8, 74. [Google Scholar] [CrossRef]
- Espigolan, R.; Baldi, F.; Boligon, A.A.; Souza, F.R.; Gordo, D.G.; Tonussi1, R.L.; Cardoso, D.F.; Oliveira, H.N.; Tonhati, H.; Sargolzaei, M.; et al. Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genom. 2013, 14, 305. [Google Scholar] [CrossRef]
- Mészáros, G. Genomic descriptors of biodiversity—A review. J. Land Manag. Food Environ. 2018, 69, 73–83. [Google Scholar] [CrossRef]
- Shin, D.; Won, K.H.; Kim, S.H.; Kim, Y.M. Extent of linkage disequilibrium and effective population size of Korean. Asian-Australas. J. Anim. Sci. 2018, 31, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, N.; Collins, A.; Xu, C.F.; McCarthy, L.C.; Hewett, D.R.; Tapper, W.; Ennis, S.; Ke, X.; Morton, N.E. The first linkage disequilibrium (LD) maps: Delineation of hot and cold blocks by diplotype analysis. Proc. Natl. Acad. Sci. USA 2002, 99, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Rougemont, Q.; Bernatchez, L. The demographic history of Atlantic salmon (Salmo salar) across its distribution range reconstructed from approximate Bayesian computations. Evolution 2018, 72, 1261–1277. [Google Scholar] [CrossRef] [PubMed]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of selection signature provides a glimpse into important economic traits in goats (Capra hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Li, X.; Han, W.; Yang, K.; Wang, H.; Zhang, Y.; Su, R.; Liu, Z.; Wang, R.; et al. Saudi Journal of Biological Sciences High-throughput sequencing of hair follicle development-related micrornas in cashmere goat at various fetal periods. Saudi J. Biol. Sci. 2018, 25, 1494–1508. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed Name | Sample Code | Sample Size (n) | Province |
|---|---|---|---|
| Nanjiang | NJ | 23 | Xinjiang |
| Qingeda | QGD | 24 | Xinjiang |
| Arbas Cashmere | AC | 59 | Inner Mongolia |
| Jining Grey | JN | 39 | Shandong |
| Louping | LP | 24 | Yunnan |
| Guangfeng | GF | 24 | Jiangxi |
| Breed Name | Code | MAF < 0.05 |
|---|---|---|
| Nanjiang | NJ | 0.284 |
| Qingeda | QGD | 0.308 |
| Arbas Cashmere | AC | 0.276 |
| Jining Grey | JN | 0.416 |
| Louping | LP | 0.353 |
| Guangfeng | GF | 0.366 |
| CHR | SNP (n) | Size (Mb) | D’ | r2 | FIS | MAF | Distance (Mb) |
|---|---|---|---|---|---|---|---|
| 1 | 2916 | 157.4 | 0.225 | 0.023 | 0.072 | 0.324 | 0.260 |
| 2 | 2502 | 136.51 | 0.225 | 0.023 | 0.075 | 0.327 | 0.260 |
| 3 | 2109 | 120.04 | 0.233 | 0.022 | 0.079 | 0.316 | 0.259 |
| 4 | 2190 | 120.74 | 0.229 | 0.023 | 0.064 | 0.319 | 0.259 |
| 5 | 2027 | 119.02 | 0.229 | 0.022 | 0.072 | 0.318 | 0.259 |
| 6 | 2161 | 117.64 | 0.235 | 0.023 | 0.073 | 0.318 | 0.259 |
| 7 | 1981 | 108.43 | 0.237 | 0.024 | 0.069 | 0.320 | 0.260 |
| 8 | 2064 | 112.67 | 0.226 | 0.022 | 0.085 | 0.321 | 0.259 |
| 9 | 1706 | 91.57 | 0.234 | 0.022 | 0.073 | 0.315 | 0.259 |
| 10 | 1846 | 101.09 | 0.223 | 0.021 | 0.079 | 0.319 | 0.259 |
| 11 | 1914 | 106.23 | 0.234 | 0.023 | 0.073 | 0.314 | 0.259 |
| 12 | 1543 | 87.28 | 0.229 | 0.023 | 0.073 | 0.318 | 0.259 |
| 13 | 1463 | 83.03 | 0.228 | 0.022 | 0.074 | 0.313 | 0.259 |
| 14 | 1719 | 94.67 | 0.231 | 0.022 | 0.080 | 0.312 | 0.260 |
| 15 | 1470 | 81.90 | 0.227 | 0.022 | 0.068 | 0.312 | 0.260 |
| 16 | 1406 | 79.37 | 0.237 | 0.024 | 0.083 | 0.312 | 0.260 |
| 17 | 1275 | 71.14 | 0.234 | 0.023 | 0.081 | 0.315 | 0.259 |
| 18 | 1165 | 67.28 | 0.223 | 0.021 | 0.070 | 0.318 | 0.259 |
| 19 | 1074 | 62.52 | 0.229 | 0.023 | 0.078 | 0.318 | 0.260 |
| 20 | 1362 | 71.78 | 0.224 | 0.022 | 0.072 | 0.320 | 0.259 |
| 21 | 1271 | 69.43 | 0.225 | 0.022 | 0.084 | 0.317 | 0.258 |
| 22 | 1040 | 60.28 | 0.231 | 0.023 | 0.077 | 0.317 | 0.259 |
| 23 | 922 | 48.87 | 0.212 | 0.020 | 0.064 | 0.321 | 0.259 |
| 24 | 1171 | 62.31 | 0.239 | 0.024 | 0.070 | 0.317 | 0.260 |
| 25 | 777 | 42.86 | 0.232 | 0.021 | 0.077 | 0.309 | 0.258 |
| 26 | 920 | 51.42 | 0.223 | 0.022 | 0.070 | 0.324 | 0.258 |
| 27 | 828 | 44.71 | 0.218 | 0.020 | 0.084 | 0.319 | 0.259 |
| 28 | 822 | 44.67 | 0.224 | 0.022 | 0.082 | 0.316 | 0.260 |
| 29 | 876 | 51.33 | 0.227 | 0.023 | 0.074 | 0.316 | 0.260 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berihulay, H.; Islam, R.; Jiang, L.; Ma, Y. Genome-Wide Linkage Disequilibrium and the Extent of Effective Population Sizes in Six Chinese Goat Populations Using a 50K Single Nucleotide Polymorphism Panel. Animals 2019, 9, 350. https://doi.org/10.3390/ani9060350
Berihulay H, Islam R, Jiang L, Ma Y. Genome-Wide Linkage Disequilibrium and the Extent of Effective Population Sizes in Six Chinese Goat Populations Using a 50K Single Nucleotide Polymorphism Panel. Animals. 2019; 9(6):350. https://doi.org/10.3390/ani9060350
Chicago/Turabian StyleBerihulay, Haile, Rabiul Islam, Lin Jiang, and Yuehui Ma. 2019. "Genome-Wide Linkage Disequilibrium and the Extent of Effective Population Sizes in Six Chinese Goat Populations Using a 50K Single Nucleotide Polymorphism Panel" Animals 9, no. 6: 350. https://doi.org/10.3390/ani9060350
APA StyleBerihulay, H., Islam, R., Jiang, L., & Ma, Y. (2019). Genome-Wide Linkage Disequilibrium and the Extent of Effective Population Sizes in Six Chinese Goat Populations Using a 50K Single Nucleotide Polymorphism Panel. Animals, 9(6), 350. https://doi.org/10.3390/ani9060350