





Pathogens 2025, 14(8), 758; https://doi.org/10.3390/pathogens14080758 (registering DOI) - 31 Jul 2025
Abstract
►
Show Figures
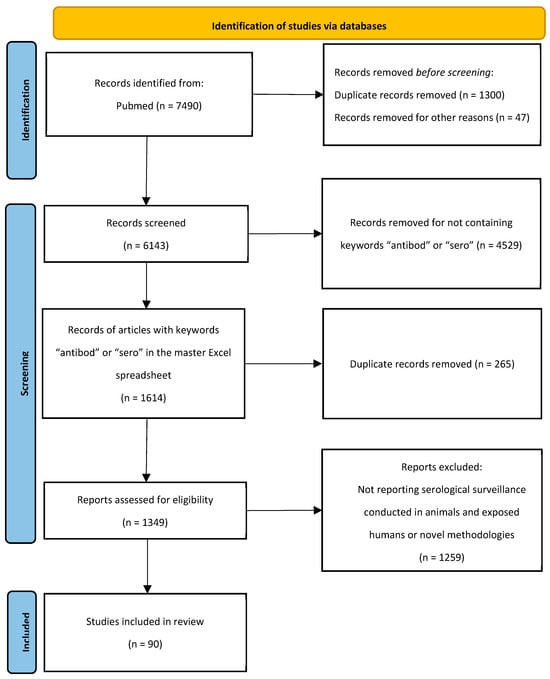
Background: COVID-19 is associated with coagulopathy and increased mortality. The ABO blood group system has been implicated in modulating susceptibility to SARS-CoV-2 infection and disease severity, but its relationship with viral RNAemia, spike gene mutations, and thrombosis remains underexplored. Methods: We analyzed 446
[...] Read more.
Background: COVID-19 is associated with coagulopathy and increased mortality. The ABO blood group system has been implicated in modulating susceptibility to SARS-CoV-2 infection and disease severity, but its relationship with viral RNAemia, spike gene mutations, and thrombosis remains underexplored. Methods: We analyzed 446 hospitalized COVID-19 patients between 2021 and 2022. SARS-CoV-2 RNAemia was assessed via RT-qPCR on whole blood, and spike gene mutations were identified through whole-genome sequencing in RNAemia-positive samples. ABO blood groups were determined by agglutination testing, and thrombotic events were evaluated using coagulation markers. Statistical analyses included chi-square tests and Kruskal–Wallis tests, with significance set at p < 0.05. Results: RNAemia was detected in 26.9% of patients, with no significant association with ABO blood group (p = 0.175). Omicron was the predominant variant, especially in blood group A (62.5%). The N501Y mutation was the most prevalent in group O (53.2%), and K417N was most prevalent in group B (36.9%), though neither reached statistical significance. Thrombotic events were significantly more common in blood group A (OR = 2.08, 95% CI = 1.3–3.4, p = 0.002), particularly among RNAemia-positive patients. Conclusions: ABO blood group phenotypes, particularly group A, may influence thrombotic risk in the context of SARS-CoV-2 RNAemia. While no direct association was found between blood group and RNAemia or spike mutations, the observed trends suggest potential host–pathogen interactions. Integrating ABO typing and RNAemia screening may enhance risk stratification and guide targeted thromboprophylaxis in COVID-19 patients.
Full article

Figure 1




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}