
Recombinants Are the Key Drivers of Recent PRRSV-2 Evolution

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. PRRSV Isolates
2.2. RNA Extraction and cDNA Synthesis
2.3. Library Preparation and Sequencing
2.4. Assembly, Annotation, and Phylogenetic Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FMDV | Foot-and-Mouth Disease Virus; |
| GP5 | Glycoprotein 5; |
| IAV | Influenza A Virus; |
| ISU | Iowa State University; |
| MAFFT | Multiple Alignment using Fast Fourier Transform; |
| NCBI | National Center for Biotechnology Information; |
| NSP | Non-Structural Protein; |
| ORF | Open Reading Frame; |
| PCR | Polymerase Chain Reaction; |
| PEDV | Porcine Epidemic Diarrhea Virus; |
| PRRSV-2 | Porcine Reproductive and Respiratory Syndrome Virus Type 2; |
| RDP5 | Recombination Detection Program version 5; |
| RFLP | Restriction Fragment Length Polymorphism; |
| RNA | Ribonucleic Acid; |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2; |
| SRA | Sequence Read Archive; |
| TSO | Template Switching Oligonucleotide. |
Appendix A

{kind=link}
{kind=link}
| Binding Region | Direction | Sequence | Length | Final Conc. (µM) |
|---|---|---|---|---|
| 3′UTR | Reverse | 5′-CCCTAATTGAATAGGTGACTTAG-3′ | 23 | 10 |
| ORF5 | Reverse | 5′-CGGANCCNTCNAGCACNACT-3′ | 20 | 10 |
| ORF4 | Reverse | 5′-CCNTTTTCNCTCATNTCNG-3′ | 19 | 10 |
| ORF2/ORF3 | Reverse | 5′-AGNTCGAANGANAANTTGCCCCT-3′ | 23 | 10 |
| ORF1b | Reverse | 5′-GTATCCGANTCNAACCCCCA-3′ | 20 | 10 |
| ORF1b | Reverse | 5′-GCCTGNNCAACCGTGCANTC-3′ | 20 | 10 |
| ORF1b | Reverse | 5′-ACNGGNGTNTGNAGCTCCTT-3′ | 20 | 10 |
| ORF1b | Reverse | 5′-GNCCACAGCGGGTCAAGCC-3′ | 19 | 10 |
| ORF1a | Reverse | 5′-GCNAANGCTTCAAGNTTNG-3′ | 19 | 10 |
| ORF1a | Reverse | 5′-GTCCANGNNTGNCCCATCAT-3′ | 20 | 10 |
| ORF1a | Reverse | 5′-ATCAANAAAAACACNANCCA-3′ | 20 | 10 |
| ORF1a | Reverse | 5′-ANACAAGANCCCCANCACTT-3’ | 20 | 10 |
| ORF1a | Reverse | 5’-GGNGGNGGNGTNTCGAGTATCA-3′ | 22 | 10 |
| ORF1a | Reverse | 5′-CCNGCNCCNTACCANTTG-3′ | 18 | 10 |
References
- Fiers, J.; Cay, A.B.; Maes, D.; Tignon, M. A comprehensive review on porcine reproductive and respiratory syndrome virus with emphasis on immunity. Vaccines 2024, 12, 942. [Google Scholar] [CrossRef]
- Kappes, M.A.; Faaberg, K.S. PRRSV structure, replication and recombination: Origin of phenotype and genotype diversity. Virology 2015, 479–480, 475–486. [Google Scholar] [CrossRef]
- Martín-Valls, G.E.; Kvisgaard, L.K.; Tello, M.; Darwich, L.; Cortey, M.; Burgara-Estrella, A.J.; Hernández, J.; Larsen, L.E.; Mateu, E. Analysis of ORF5 and full-length genome sequences of porcine reproductive and respiratory syndrome virus isolates of genotypes 1 and 2 retrieved worldwide provides evidence that recombination is a common phenomenon and may produce mosaic isolates. J. Virol. 2014, 88, 3170–3181. [Google Scholar] [CrossRef]
- Paploski, I.A.D.; Pamornchainavakul, N.; Makau, D.N.; Rovira, A.; Corzo, C.A.; Schroeder, D.C.; Cheeran, M.C.-J.; Doeschl-Wilson, A.; Kao, R.R.; Lycett, S.; et al. Phylogenetic structure and sequential dominance of sub-lineages of PRRSV type-2 lineage 1 in the United States. Vaccines 2021, 9, 608. [Google Scholar] [CrossRef] [PubMed]
- Yim-im, W.; Anderson, T.K.; Paploski, I.A.D.; VanderWaal, K.; Gauger, P.; Krueger, K.; Shi, M.; Main, R.; Zhang, J. Refining PRRSV-2 genetic classification based on global ORF5 sequences and investigation of their geographic distributions and temporal changes. Microbiol. Spectr. 2023, 11, e02916–e02923. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lin, S.; Quan, D.; Wang, H.; Huang, J.; Wang, Y.; Ren, T.; Ouyang, K.; Chen, Y.; Huang, W.; et al. Full genomic analysis of new variants of porcine reproductive and respiratory syndrome virus revealed multiple recombination events between different lineages and sublineages. Front. Vet. Sci. 2020, 7, 603. [Google Scholar] [CrossRef] [PubMed]
- Pamornchainavakul, N.; Kikuti, M.; Paploski, I.A.D.; Makau, D.N.; Rovira, A.; Corzo, C.A.; VanderWaal, K. Measuring how recombination reshapes the evolutionary history of PRRSV-2: A genome-based phylodynamic analysis of the emergence of a novel PRRSV-2 variant. Front. Vet. Sci. 2022, 9, 846904. [Google Scholar] [CrossRef]
- Schroeder, D.C.; Odogwu, N.M.; Kevill, J.; Yang, M.; Krishna, V.D.; Kikuti, M.; Pamornchainavakul, N.; Vilalta, C.; Sanhueza, J.; Corzo, C.A.; et al. Phylogenetically distinct near-complete genome sequences of porcine reproductive and respiratory syndrome virus type 2 variants from four distinct disease outbreaks at U.S. swine farms over the past 6 years. Microbiol. Resour. Announc. 2021, 10, e00260-21. [Google Scholar] [CrossRef]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retroviruses 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved algorithmic complexity for the 3SEQ recombination detection algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Iowa State University Veterinary Diagnostic Laboratory. ISU PRRSView Web Tool. Available online: https://prrsv.vdl.iastate.edu/ (accessed on 22 January 2025).
- Wesley, R.D.; Mengeling, W.L.; Lager, K.M.; Clouser, D.F.; Landgraf, J.G.; Frey, M.L. Differentiation of a porcine reproductive and respiratory syndrome virus vaccine strain from North American field strains by restriction fragment length polymorphism analysis of ORF 5. J. Vet. Diagn. Investig. 1998, 10, 140–144. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef]
- Cui, X.-Y.; Xia, D.-S.; Luo, L.-Z.; An, T.-Q. Recombination of porcine reproductive and respiratory syndrome virus: Features, possible mechanisms, and future directions. Viruses 2024, 16, 929. [Google Scholar] [CrossRef]
- Zhao, K.; Ye, C.; Chang, X.B.; Jiang, C.G.; Wang, S.J.; Cai, X.H.; Tong, G.Z.; Tian, Z.J.; Shi, M.; An, T.Q. Importation and recombination are responsible for the latest emergence of highly pathogenic porcine reproductive and respiratory syndrome virus in China. J. Virol. 2015, 89, 10712–10716. [Google Scholar] [CrossRef]
- Wang, A.; Chen, Q.; Wang, L.; Madson, D.; Harmon, K.; Gauger, P.; Zhang, J.; Li, G. Recombination between vaccine and field strains of porcine reproductive and respiratory syndrome virus. Emerg. Infect. Dis. 2019, 25, 2335–2337. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Kang, R.; Yu, J.; Xie, B.; Chen, C.; Li, X.; Xie, J.; Ye, Y.; Xiao, L.; Zhang, J.; et al. Genetic characterization and pathogenicity of a novel recombined porcine reproductive and respiratory syndrome virus 2 among NADC30-like, JXA1-like, and MLV-like strains. Viruses 2018, 10, 551. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Xia, D.; Huang, X.; Sun, Y.; Shi, M.; Zhang, J.; Li, G.; Yang, Y.; Wang, H.; Cai, X.; et al. Analysis of recombinant characteristics based on 949 PRRSV-2 genomic sequences obtained from 1991 to 2021 shows that viral multiplication ability contributes to dominant recombination. Microbiol. Spectr. 2022, 10, e0293422. [Google Scholar] [CrossRef] [PubMed]
- Abd El Rahman, S.; Hoffmann, B.; Karam, R.; El-Beskawy, M.; Hamed, M.F.; Forth, L.F.; Höper, D.; Eschbaumer, M. Sequence analysis of Egyptian foot-and-mouth disease virus field and vaccine strains: Intertypic recombination and evidence for accidental release of virulent virus. Viruses 2020, 12, 990. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Liu, Y.; Chen, Y.; Jiao, W.; Feng, H.; Wei, Q.; Wang, J.; Zhang, Y.; Zhang, G. Isolation and identification of a recombinant porcine epidemic diarrhea virus with a novel insertion in S1 domain. Front. Microbiol. 2021, 12, 667084. [Google Scholar] [CrossRef]
- Peng, Q.; Fu, P.; Zhou, Y.; Lang, Y.; Zhao, S.; Wen, Y.; Wang, Y.; Wu, R.; Zhao, Q.; Du, S.; et al. Phylogenetic analysis of porcine epidemic diarrhea virus (PEDV) during 2020–2022 and isolation of a variant recombinant PEDV strain. Int. J. Mol. Sci. 2024, 25, 10878. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Recombination in coronaviruses, with a focus on SARS-CoV-2. Viruses 2022, 14, 1239. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.Á.; Filip, I.; Chowdhury, R.; Maranas, C.D.; Sorger, P.K.; AlQuraishi, M.; Rabadan, R. Recombination and lineage-specific mutations linked to the emergence of SARS-CoV-2. Genome Med. 2021, 13, 124. [Google Scholar] [CrossRef]
- Pipek, O.A.; Medgyes-Horváth, A.; Stéger, J.; Papp, K.; Visontai, D.; Koopmans, M.; Nieuwenhuijse, D.; Oude Munnink, B.B.; VEO Technical Working Group; Csabai, I. Systematic detection of co-infection and intra-host recombination in more than 2 million global SARS-CoV-2 samples. Nat. Commun. 2024, 15, 517. [Google Scholar] [CrossRef]
- Zimmer, S.M.; Burke, D.S. Historical perspective–emergence of influenza A (H1N1) viruses. N. Engl. J. Med. 2009, 361, 279–285. [Google Scholar] [CrossRef]
- Li, C.; Culhane, M.R.; Schroeder, D.C.; Cheeran, M.C.-J.; Galina Pantoja, L.; Jansen, M.L.; Torremorell, M. Vaccination decreases the risk of influenza A virus reassortment but not genetic variation in pigs. eLife 2022, 11, e78618. [Google Scholar] [CrossRef]
- Perfumo, C.J.; Pereda, A.; Jongkaewwattana, A.; Chen, Z.; Perez, D.R.; Ma, J. Editorial: Emerging swine viruses. Front. Vet. Sci. 2020, 7, 132. [Google Scholar] [CrossRef]
- Wei, C.; Liu, C.; Chen, G.; Yang, Y.; Li, J.; Dan, H.; Dai, A.; Huang, C.; Luo, M.; Liu, J. Genetic characterization and pathogenicity of two recombinant PRRSV-2 strains from lineages 1, 3, 5, and 8 emerged in China. BMC Vet. Res. 2025, 21, 341. [Google Scholar] [CrossRef]
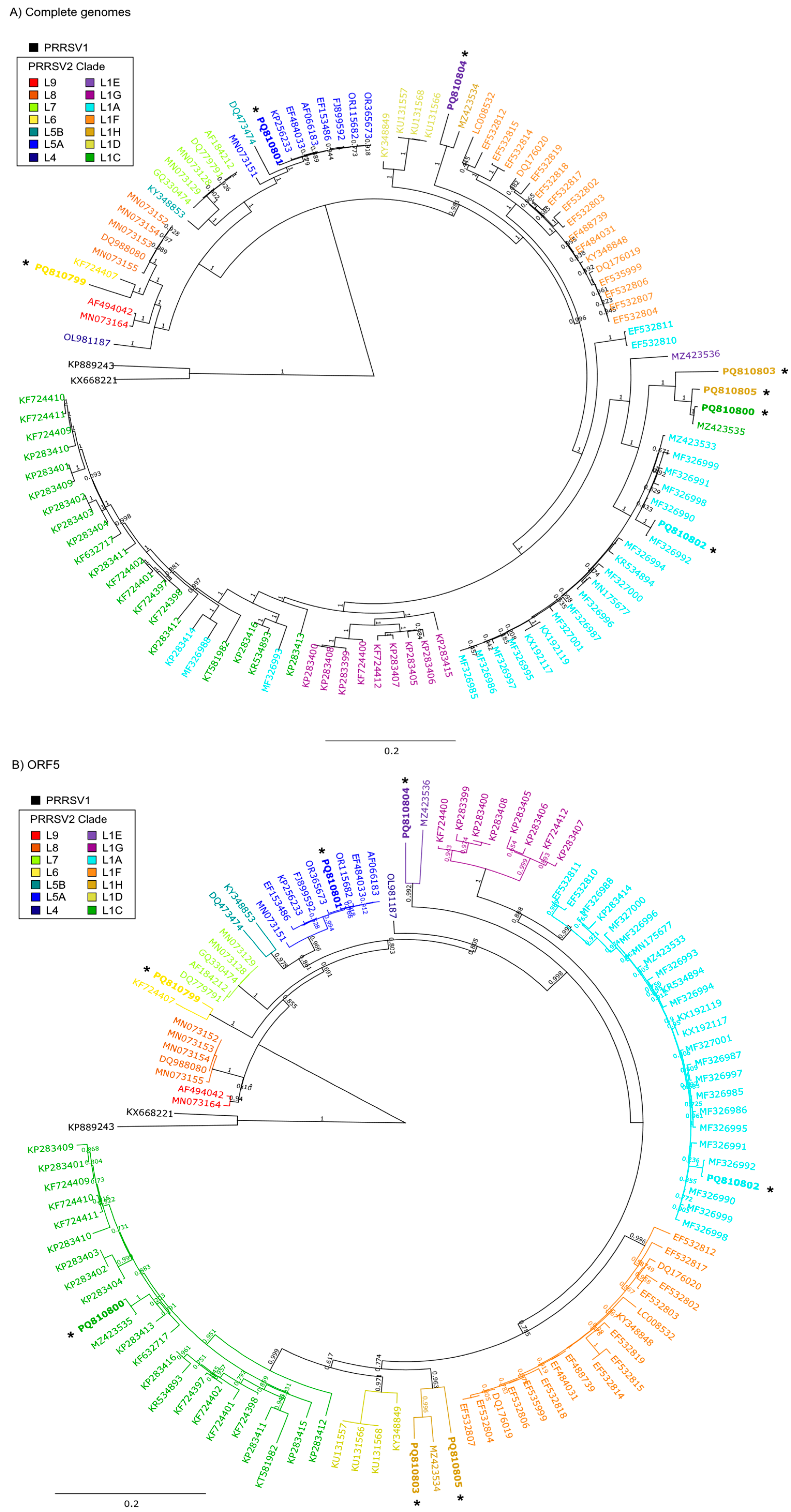
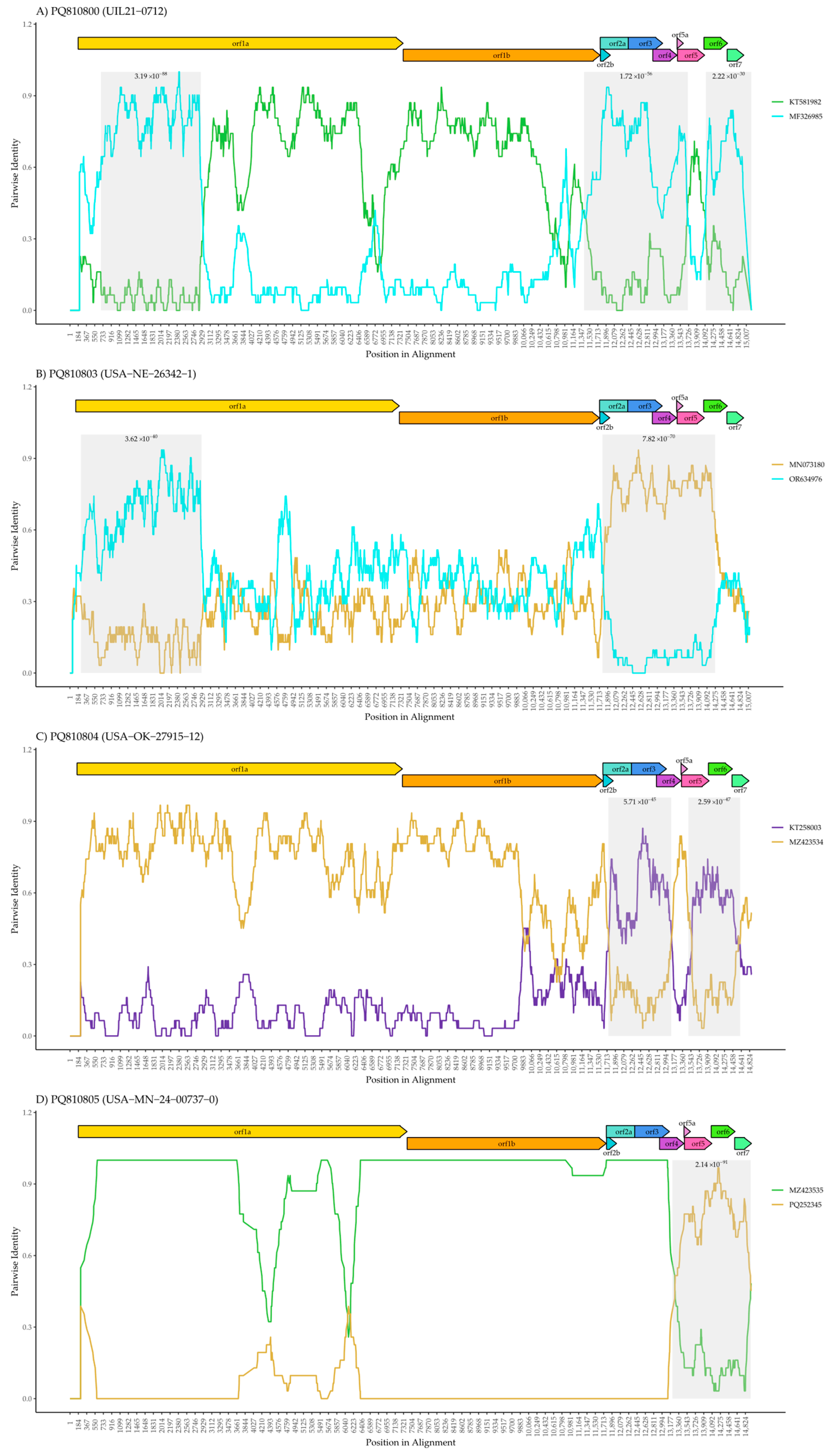
| Accession Number | BioSample | SRA Accession | Isolate | Collection Date | State | Reads Mapped (bp) | Genome Length (bp) |
|---|---|---|---|---|---|---|---|
| PQ810799 | SAMN45150044 | SRR31590011 | KS2006-72109 1-3-4 L6 | 2006 | Kansas | 143,295 | 15,397 |
| PQ810800 | SAMN45150045 | SRR31590010 | UIL21-0712 1-4-4 L1C | 2021 | Illinois | 60,642 | 15,119 |
| PQ810801 | SAMN45150046 | SRR31590009 | USA-IL-23295-GA 2-5-2 L5A | 2022 | Illinois | 116,314 | 14,642 |
| PQ810802 | SAMN45150047 | SRR31590008 | USA-IN-65239-GA 1-7-4 L1A | 2014 | Indiana | 93,025 | 15,059 |
| PQ810803 | SAMN45150048 | SRR31590007 | USA-NE-26342-1 1-8-4 L1H | 2022 | Nebraska | 4365 | 15,067 |
| PQ810804 | SAMN45150049 | SRR31590006 | USA-OK-27915-12 1-4-2 L1E | 2022 | Oklahoma | 6207 | 14,915 |
| PQ810805 | SAMN45150050 | SRR31590005 | USA-MN-24-00737-0 1-12-2 L1H | 2024 | Minnesota | 2042 | 15,125 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellegrini Ferreira, C.; Galina-Pantoja, L.; Wagner, M.; Schroeder, D.C. Recombinants Are the Key Drivers of Recent PRRSV-2 Evolution. Pathogens 2025, 14, 743. https://doi.org/10.3390/pathogens14080743
Pellegrini Ferreira C, Galina-Pantoja L, Wagner M, Schroeder DC. Recombinants Are the Key Drivers of Recent PRRSV-2 Evolution. Pathogens. 2025; 14(8):743. https://doi.org/10.3390/pathogens14080743
Chicago/Turabian StylePellegrini Ferreira, Clarissa, Lucina Galina-Pantoja, Mark Wagner, and Declan C. Schroeder. 2025. "Recombinants Are the Key Drivers of Recent PRRSV-2 Evolution" Pathogens 14, no. 8: 743. https://doi.org/10.3390/pathogens14080743
APA StylePellegrini Ferreira, C., Galina-Pantoja, L., Wagner, M., & Schroeder, D. C. (2025). Recombinants Are the Key Drivers of Recent PRRSV-2 Evolution. Pathogens, 14(8), 743. https://doi.org/10.3390/pathogens14080743