
Phylogenetic Reclassification of Metarhizium granulomatis and Metarhizium viride Species Complex


Abstract
1. Introduction
2. Materials and Methods
2.1. Fungal Isolates
2.2. PCR and DNA Sequencing
2.3. Analyzing Data According to Phylogeny
3. Results
3.1. RNA Polymerase II Second Largest Subunit (NRPB2) of M. granulomatis
3.2. RNA Polymerase II Second Largest Subunit (NRPB2) of M. viride
3.3. Translation Elongation Factor 1 alpha (EF1α)
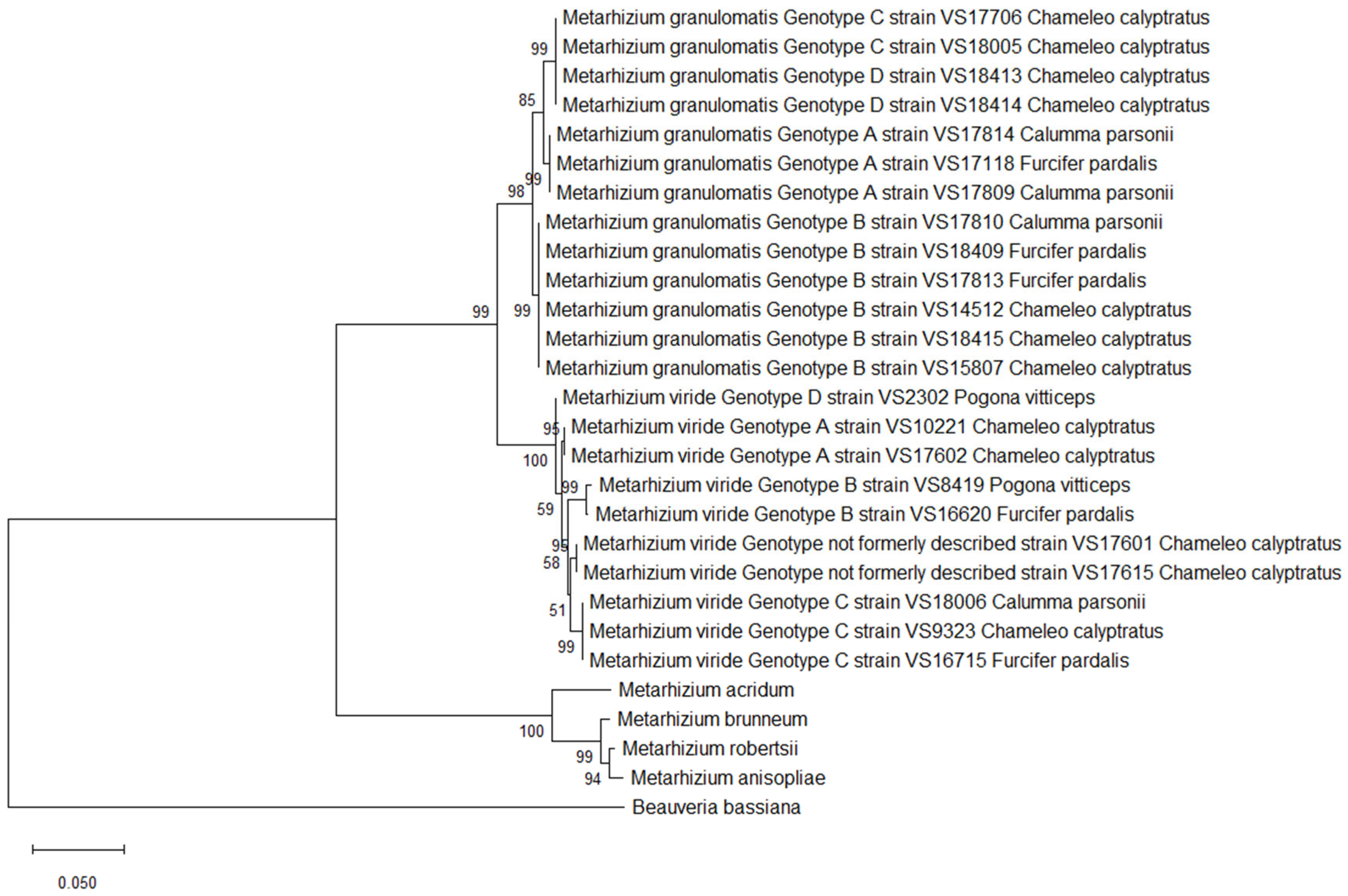
3.4. Multilocus Analysis
3.5. Isolates and Associated Findings
4. Discussion
4.1. Phylogenetic Analysis of M. granulomatis Using a Multilocus Approach
4.2. Phylogenetic Analysis of M. viride Using a Multilocus Approach
4.3. Evaluation of the Phylogenetic Analysis Using NRPB2 (RPB1)
4.4. Evaluation of the Phylogenetic Analysis Using NRPB2 (RPB2)
4.5. Evaluation of the Phylogenetic Analysis Using EF1α
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PCR | Polymerase chain reaction |
| SNP | Single-nucleotide polymorphism |
| CDS | Coding DNA sequence |
| NRPB2 | RNA polymerase II second largest subunit |
| RPB1 | First analyzed locus of the RNA polymerase II second largest subunit |
| RPB2 | Second analyzed locus RNA polymerase II second largest subunit |
| EF1α | Translation elongation factor 1 alpha |
References
- Sigler, L.; Gibas, C.F.C.; Kokotovic, B.; Bertelsen, M.F. Disseminated mycosis in veiled chameleons (Chamaeleo calyptratus) caused by Chamaeleomyces granulomatis, a new fungus related to Paecilomyces viridis. J. Clin. Microbiol. 2010, 48, 3182–3192. [Google Scholar] [CrossRef]
- Schmidt, V.; Klasen, L.; Schneider, J.; Hübel, J.; Cramer, K. Pulmonary fungal granulomas and fibrinous pneumonia caused by different hypocrealean fungi in reptiles. Vet. Microbiol. 2018, 225, 58–63. [Google Scholar] [CrossRef]
- Horgan, M.D.; Alexander, A.B.; Innis, C.; Stacy, B.A.; Gai, J.J.; Pesavento, P.A.; Highland, M.A.; Liguori, B.L.; Norton, T.M.; Wellehan, J.F.X.; et al. Pulmonary and coelomic mycoses due to Metarhizium and Beauveria species in reptiles. J. Zoo. Wildl. Med. 2022, 53, 605–612. [Google Scholar] [CrossRef]
- Warwick, C. Observations on disease-associated preferred body temperatures in reptiles. Appl. Anim. Behav. Sci. 1991, 28, 375–380. [Google Scholar] [CrossRef]
- Pough, F.H. Recommendations for the care of amphibians and reptiles in academic institutions. ILAR J. 1991, 33, S1–S21. [Google Scholar] [CrossRef]
- Schmidt, V.; Klasen, L.; Schneider, J.; Hübel, J.; Pees, M. Fungal dermatitis, glossitis and disseminated visceral mycosis caused by different Metarhizium granulomatis genotypes in veiled chameleons (Chamaeleo calyptratus) and first isolation in healthy lizards. Vet. Microbiol. 2017, 207, 74–82. [Google Scholar] [CrossRef]
- Schmidt, V.; Klasen, L.; Schneider, J.; Hübel, J.; Pees, M. Characterization of Metarhizium viride mycosis in veiled chameleons (Chamaeleo calyptratus), panther chameleons (Furcifer pardalis), and inland bearded dragons (Pogona vitticeps). J. Clin. Microbiol. 2017, 55, 832–843. [Google Scholar] [CrossRef]
- Klasen, L.; Pees, M.; Schmidt, V. Simultaneous detection of Metarhizium viride, Metarhizium granulomatis and Metarhizium anisopliae species complex in veiled chameleons (Chamaeleo calyptratus), panther chameleon (Furcifer pardalis) and a central bearded dragon (Pogona vitticeps). Berl. Münchener Tierärztliche Wochenschrift 2019, 132, 428–434. [Google Scholar] [CrossRef]
- Kepler, R.M.; Luangsa-ard, J.J.; Hywel-Jones, N.L.; Quandt, C.A.; Sung, G.H.; Rehner, S.A.; Aime, M.C.; Henkel, T.W.; Sanjuan, T.; Zare, R.; et al. A phylogenetically-based nomenclature for Cordycipitaceae (Hypocreales). IMA Fungus 2017, 8, 335–353. [Google Scholar] [CrossRef]
- Kepler, R.M.; Humber, R.A.; Bischoff, J.F.; Rehner, S.A. Clarification of generic and species boundaries for Metarhizium and related fungi through multigene phylogenetics. Mycologia 2014, 106, 811–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Whelen, S.; Hall, B.D. Phylogenetic relationships among ascomycetes: Evidence from an RNA polymerse II subunit. Mol. Biol. Evol. 1999, 16, 1799–1808. [Google Scholar] [CrossRef]
- Mongkolsamrit, S.; Khonsanit, A.; Thanakitpipattana, D.; Tasanathai, K.; Noisripoom, W.; Lamlertthon, S.; Himaman, W.; Houbraken, J.; Samson, R.A.; Luangsa-ard, J. Revisiting Metarhizium and the description of new species from Thailand. Stud Mycol. 2020, 95, 171–251. [Google Scholar] [CrossRef]
- Malkus, A.; Linda Chang, P.F.; Zuzga, S.M.; Chung Kren Shao, J.; Cunfer, B.M.; Arseniuk, E.; Ueng, P.P. RNA polymerase II gene (RPB2) encoding the second largest protein subunit in Phaeosphaeria nodorum and P. avenaria. Mycol. Res. 2006, 110, 1152–1164. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, J.F.; Rehner, S.A.; Humber, R.A. Metarhizium frigidum sp. nov.: A cryptic species of M. anisopliae and a member of the M. flavoviride complex. Mycologia 2006, 98, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Mülhardt, C. Der Experimentator: Molekularbiologie/Genomics; Spektrum Akademischer Verlag: Heidelberg, Germany, 2009. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. Molecular evolutionary genetics analysis version 12 for adaptive and green computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press: Cambridge, MA, USA, 1969; pp. 21–132. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Rehner, S.A.; Kepler, R.M. Species limits, phylogeography and reproductive mode in the Metarhizium anisopliae complex. J. Invertebr. Pathol. 2017, 148, 60–66. [Google Scholar] [CrossRef]
- Du, L.C. Plant Protection. South China Agricultural University: Guangzhou, China, 2021; unpublished work. [Google Scholar]
- Bischoff, J.F.; Rehner, S.A.; Humber, R.A. A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 2009, 101, 512–530. [Google Scholar] [CrossRef]
- Saud, Z.; Kortsinoglou, A.M.; Kouvelis, V.N.; Butt, T.M. Telomere length de novo assembly of all 7 chromosomes and mitogenome sequencing of the model entomopathogenic fungus, Metarhizium brunneum, by means of a novel assembly pipeline. BMC Genom. 2021, 22, 87. [Google Scholar] [CrossRef]
- Hu, X.; Xiao, G.; Zheng, P.; Shang, Y.; Su, Y.; Zhang, X.; Liu, X.; Zhan, S.; Leger, R.J.S.; Wang, C. Trajectory and genomic determinants of fungal-pathogen speciation and host adaptation. Proc. Natl. Acad. Sci. USA 2014, 111, 16796–16801. [Google Scholar] [CrossRef]
- Zhang, X. Molecular Identification of Metarhizium Strains Isolated from Pests and Soil in China. Anhui Provincial Key Laboratory for Microbial Pest Control, Anhui Agricultural University: Hefei, China, 2014; submitted. [Google Scholar]
- Nielsen, K.N.; Salgado, J.F.M.; Natsopoulou, M.E.; Kristensen, T.; Stajich, J.E.; De Fine Licht, H.H. Diploidy within a haploid genus of entomopathogenic fungi. Genome Biol. Evol. 2021, 13, evab158. [Google Scholar] [CrossRef] [PubMed]
- Inglis, G.D.; Duke, G.M.; Goettel, M.S.; Kabaluk, J.T.; Ortega-Polo, R. Biogeography and genotypic diversity of Metarhizium brunneum and Metarhizium robertsii in northwestern North American soils. Can. J. Microbiol. 2019, 65, 261–281. [Google Scholar] [CrossRef] [PubMed]
- Khonsanit, A.; Luangsa-ard, J.J.; Thanakitpipattana, D.; Noisripoom, W.; Chaitika, T.; Kobmoo, N. Cryptic diversity of the genus Beauveria with a new species from Thailand. Mycol. Prog. 2020, 19, 291–315. [Google Scholar] [CrossRef]
- Yang, Y.T.; Chen, C.Y.; Yang, Y.Y. Phylogenetic Analysis of Beauveria bassiana. Agricultural Research and Extension Station, MOA, Plant Protection Laboratory: Taoyuan City, Taiwan, 2024; submitted. [Google Scholar]
- Dalla Nora, D. Fungos Entomopatogenicos para o Controle do Percevejo da Soja. Ciencia do Solo, Universidade Federal de Santa Maria: Santa Maria, Brazil, 2019; submitted. [Google Scholar]
- Stackebrandt, E.; Ebers, J. Taxonomic parameters revisited: Tarnished gold standards. Microbiol. Today 2006, 33, 152–155. [Google Scholar]


{kind=link}
{kind=link}
| Target [References] | Forward/Reverse Primers | Annealing Temperature [°C] | Amplicon Size [bp] |
|---|---|---|---|
| RPB1 [11,12] | frPB2-5f2 (5′ GGG GWG AYC AGA AGA AGG C 3′)/ frPB2-7cR (5′ CCC ATR GCT TGY TTR CCC AT 3′) | 60 | 1000 |
| RPB2 [11,13] | fRPB2-7cF (5′ ATG GGY AAR CAA GCY ATG GG 3′)/ fRPB2-11aR (5′ GCR TGG ATC TTR TCR TCS ACC 3′) | 58 | 700 |
| EF1α [14] | 5TEF_EF1T (5′ ATG GGT AAG GAR GAC AAG AC 3′)/ 5TEF_EF2T (5′ GGA AGT ACC AGT GAT CAT GT 3′) | 58 | 800 |
| GT | Isolates (NCBI Acc.-Nr.) | Host Species | Origin of Isolation | Associated Pathological Findings and/or Additional Disease |
|---|---|---|---|---|
| G A | VS17118 a (PV231534, PV231557, PV231580) | Panther chameleon (Furcifer pardalis) | throat and cloaca | Tail tip necrosis |
| VS17809 * (PV231535, PV231558, PV231581) VS17814 a (PV231536, PV231559, PV231582) | Parson’s chameleon (Calumma parsonii) (n = 2) | cloaca (n = 2) | Obstipation, diphtheroid colorectitis; NAF | |
| G B | VS17813 a (PV231537, PV231560, PV231583) | Carpet chameleon (Furcifer lateralis) | cloaca | Petechiae on tongue |
| VS18415 *,#,a (PV231538, PV231561, PV231584), VS14512 a (PV231539, PV231562, PV231585), VS15807 (PV231540, PV231563, PV231586) | Veiled chameleon (Chameleo calyptratus) (n = 3) | throat and cloaca (n = 2), throat (n = 1) | Petechiae on tongue (n = 2), hematochezia, tongue prolapse; granulomatous mycotic pharyngitis; mycobacteriosis | |
| VS17810 * (PV231541, PV231564, PV231587) | Parson’s chameleon | cloaca | Obstipation, diphtheroid colorectitis | |
| VS18409 (PV231542, PV231565, PV231588) | Panther chameleon | cloaca | Petechiae on tongue | |
| G C | VS17706 (PV231545, PV231568, PV231591), VS18005 a (PV231546, PV231569, PV231592) | Veiled chameleon (n = 2) | rectum (n = 1), throat (n = 1) | Hematochezia, petechiae on tongue; egg yolk serositis, granulomatous mycotic colorectitis; squamous cell carcinoma |
| G D | VS18413 a (PV231543, PV231566, PV231589), VS18414 *,#,a (PV231544, PV231567, PV231590) | Veiled chameleon (n = 2) | tongue (n = 1), throat and cloaca (n = 1) | Hematochezia, petechiae on tongue, coccidiosis; tongue prolapse |
| V A | VS17602 a (PV231547, PV231570, PV231593), VS10221 (PV231548, PV231571, PV231594) | Veiled chameleon (n = 2) | tongue (n = 1), liver (n = 1) | Granulomatous mycotic glossitis; visceral mycosis |
| V B | VS8419 (PV231550, PV231573, PV231596) VS16620 a (PV231551, PV231574, PV231597) | Central bearded dragon, Panther chameleon | throat and liver, fecal sample | Granulomatous mycotic pharyngitis, visceral mycosis; NAF |
| V C | VS9323 a (PV231552, PV231575, PV231598) | Veiled chameleon | tongue | Granulomatous mycotic glossitis, tongue paralysis |
| VS16715 a (PV231553, PV231576, PV231599) | Panther chameleon | throat | NAF | |
| VS18006 (PV231554, PV231577, PV231600) | Parson’s chameleon | tongue | NAF | |
| V D | VS2302 a (PV231549, PV231572, PV231595) | Central bearded dragon (Pogona vitticeps) | throat | Granulomatous mycotic pharyngitis |
| V not formerly described | VS17601 a (PV231555, PV231578, PV231601), VS17615 a (PV231556, PV231579, PV231602) | Veiled chameleon (n = 2) | throat (n = 1); fecal sample (n = 1) | Petechiae on tongue, conjunctivitis; NAF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Würf, J.; Schmidt, V. Phylogenetic Reclassification of Metarhizium granulomatis and Metarhizium viride Species Complex. Pathogens 2025, 14, 745. https://doi.org/10.3390/pathogens14080745
Würf J, Schmidt V. Phylogenetic Reclassification of Metarhizium granulomatis and Metarhizium viride Species Complex. Pathogens. 2025; 14(8):745. https://doi.org/10.3390/pathogens14080745
Chicago/Turabian StyleWürf, Johanna, and Volker Schmidt. 2025. "Phylogenetic Reclassification of Metarhizium granulomatis and Metarhizium viride Species Complex" Pathogens 14, no. 8: 745. https://doi.org/10.3390/pathogens14080745
APA StyleWürf, J., & Schmidt, V. (2025). Phylogenetic Reclassification of Metarhizium granulomatis and Metarhizium viride Species Complex. Pathogens, 14(8), 745. https://doi.org/10.3390/pathogens14080745