


Viruses 2026, 18(5), 585; https://doi.org/10.3390/v18050585 - 21 May 2026
Abstract
Dengue virus (DENV) poses a growing risk in Tanzania, yet its genetic diversity in mosquito populations remains poorly understood. Using Nanopore sequencing, we recovered full coding sequences from six DENV-2 positive mosquito pools collected in Dar es Salaam outside recognized outbreak periods. Phylogenetic
[...] Read more.
Dengue virus (DENV) poses a growing risk in Tanzania, yet its genetic diversity in mosquito populations remains poorly understood. Using Nanopore sequencing, we recovered full coding sequences from six DENV-2 positive mosquito pools collected in Dar es Salaam outside recognized outbreak periods. Phylogenetic analysis placed these sequences in a distinct monophyletic clade within genotype II, separate from strains linked to Tanzania’s 2014 outbreak. Instead, they clustered with Asian lineages and showed the closest relatedness to DENV-2 strains from Kenya (2013) and India (2014), with divergence estimated to have occurred around 2010. Variant profiling identified 212 low-frequency intra-pool variants, predominantly non-synonymous changes in the NS3, NS4B, and NS5 coding regions. These results suggest a previously unrecognized introduction of genotype II that is now circulating silently within local mosquito populations. Our findings highlight the value of genomic surveillance in mosquito vectors for early detection of arboviral threats, even in the absence of reported human cases.
Full article
(This article belongs to the Special Issue Current Trends in Arbovirus Outbreaks and Research)
►
Show Figures

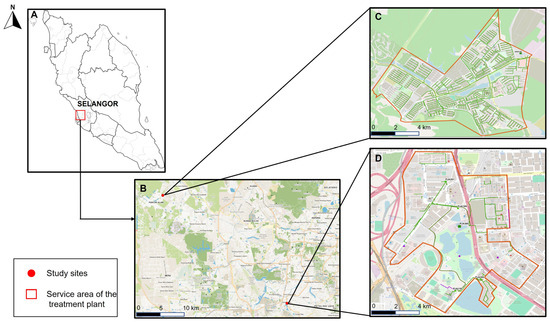
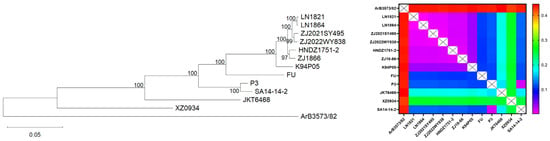
Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}