

The Role of Prion Protein in Reelin/Dab1 Signaling: Implications for Neurodegeneration
 , , and
, , and 
Abstract
1. Introduction
2. Methods
2.1. Animals
2.2. Primary Neuronal Culture Preparation
2.3. Production of Reelin-Conditioned Supernatant
2.4. Intracerebral Inoculation of Prions
2.5. Primary Neuronal Lysate Preparation
2.6. Tissue Homogenization for Protein Extraction
2.7. Biochemical Analysis of Brain Homogenates and Neuronal Lysates
2.8. Immunoprecipitation of Dab1 from Brain Homogenates
2.9. Co-Immunoprecipitation Experiments
2.10. Antibodies
2.11. RNA Extraction from Brain Samples and Real-Time PCR
2.12. Statistical Analysis
3. Results
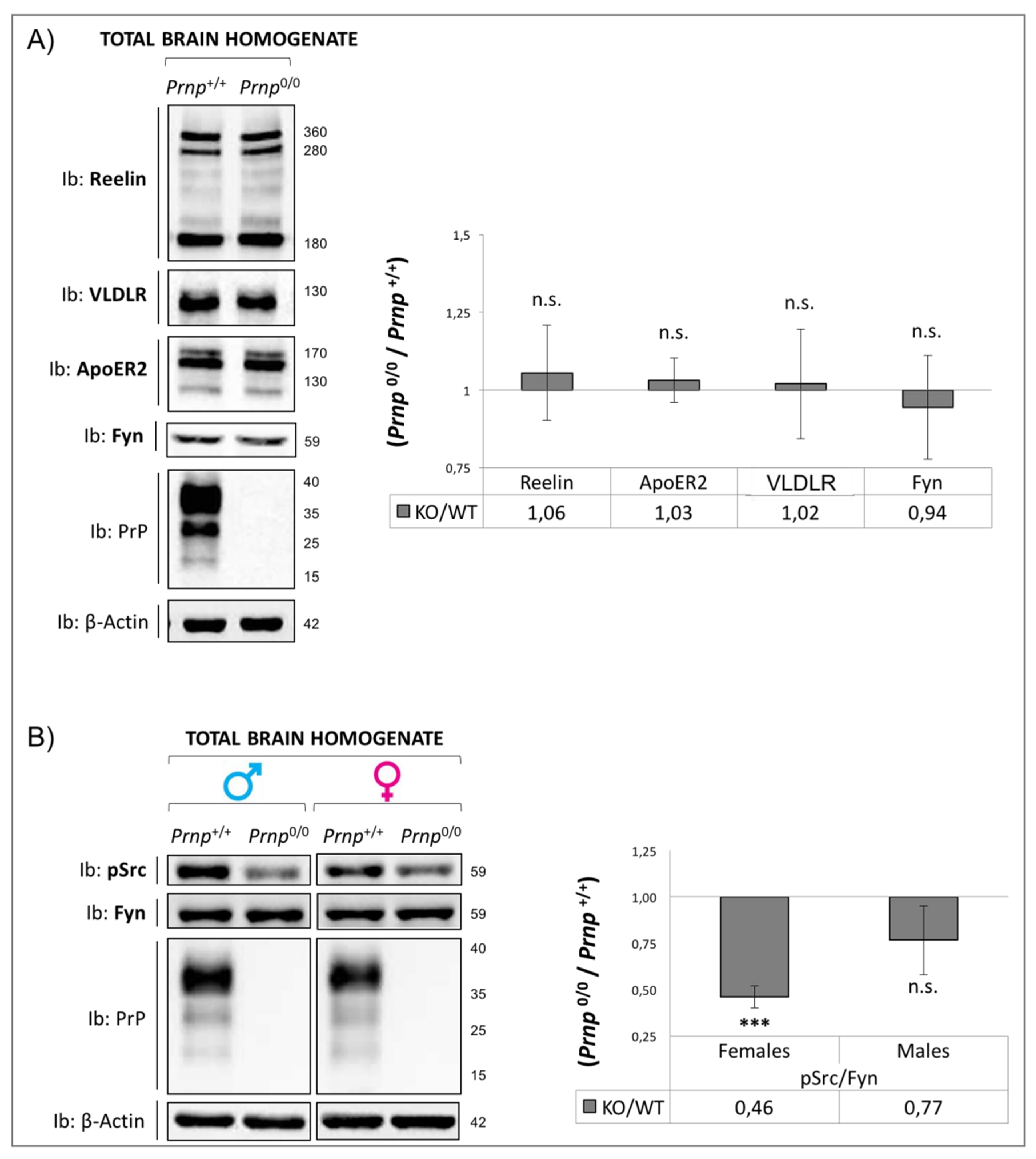
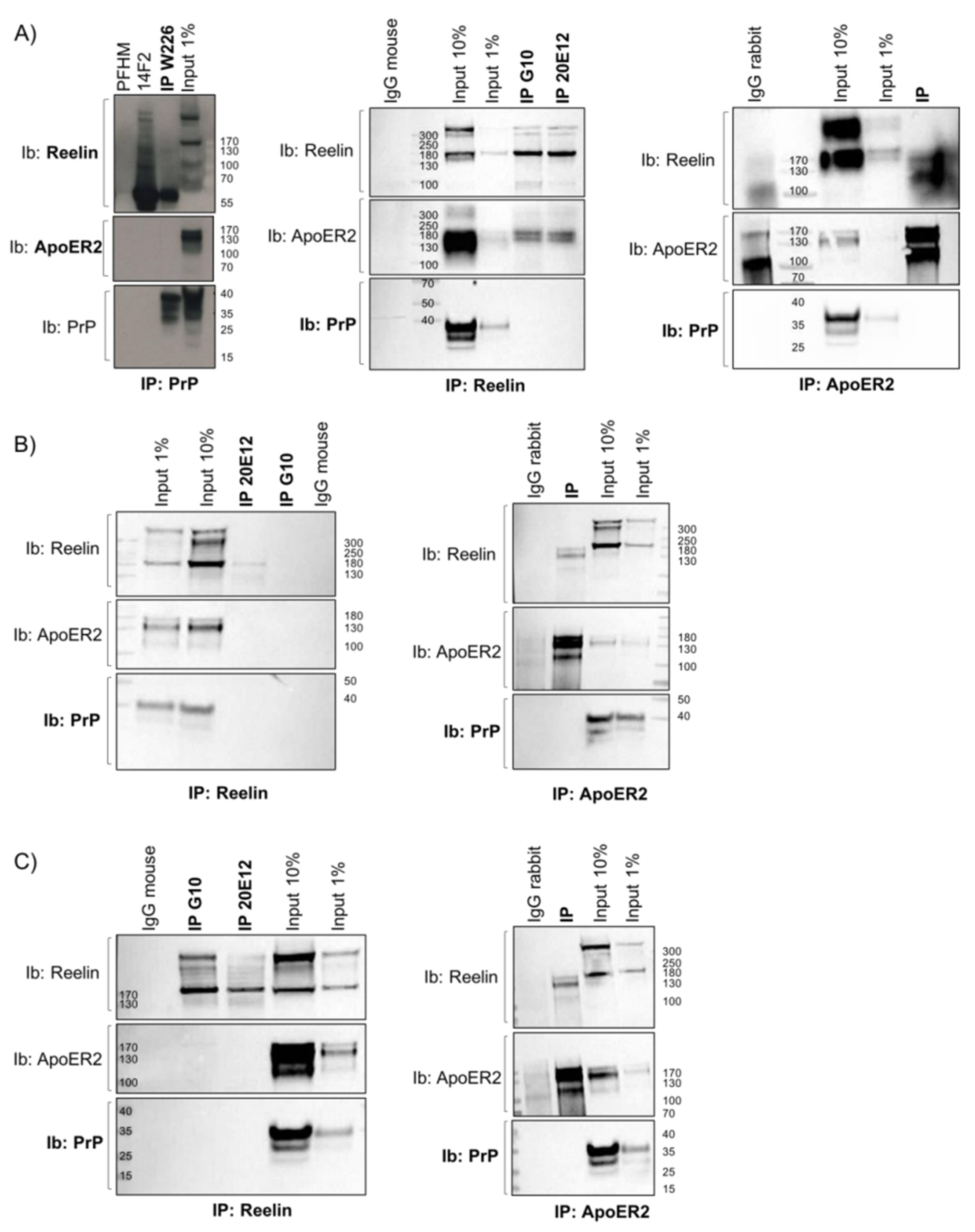
3.1. Dab1 Expression Analysis

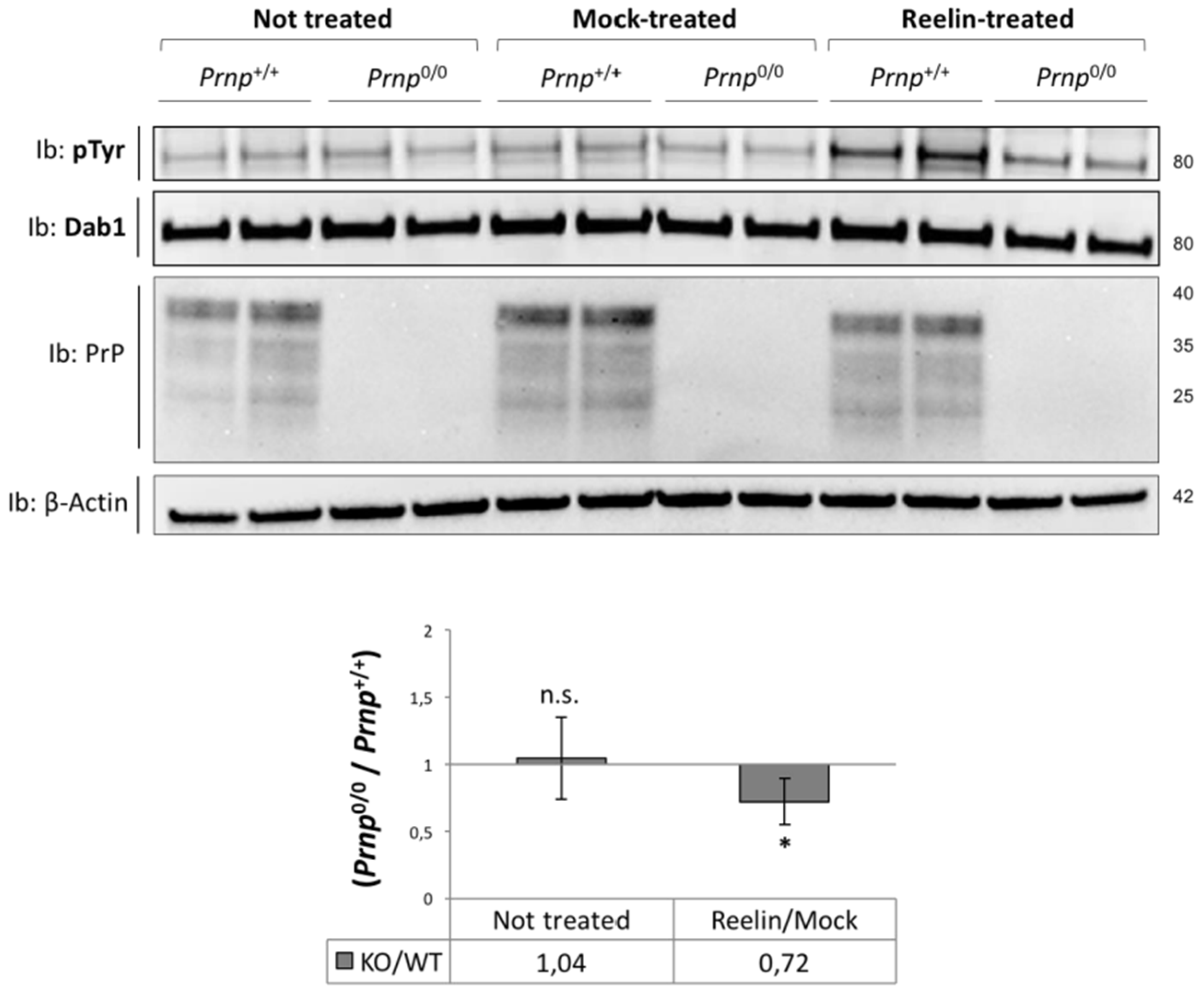
3.2. Stimulation with a Reelin-Conditioned Medium
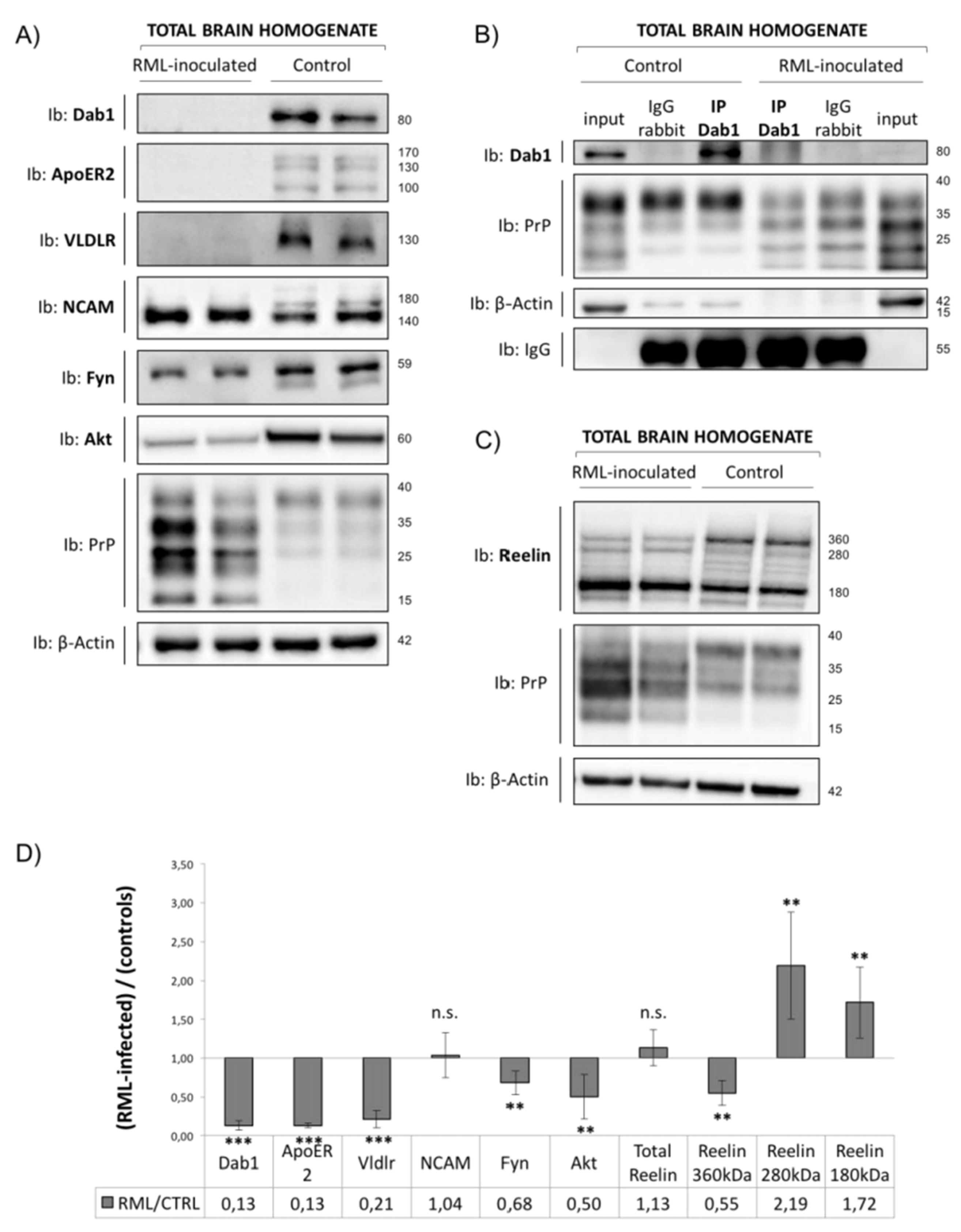
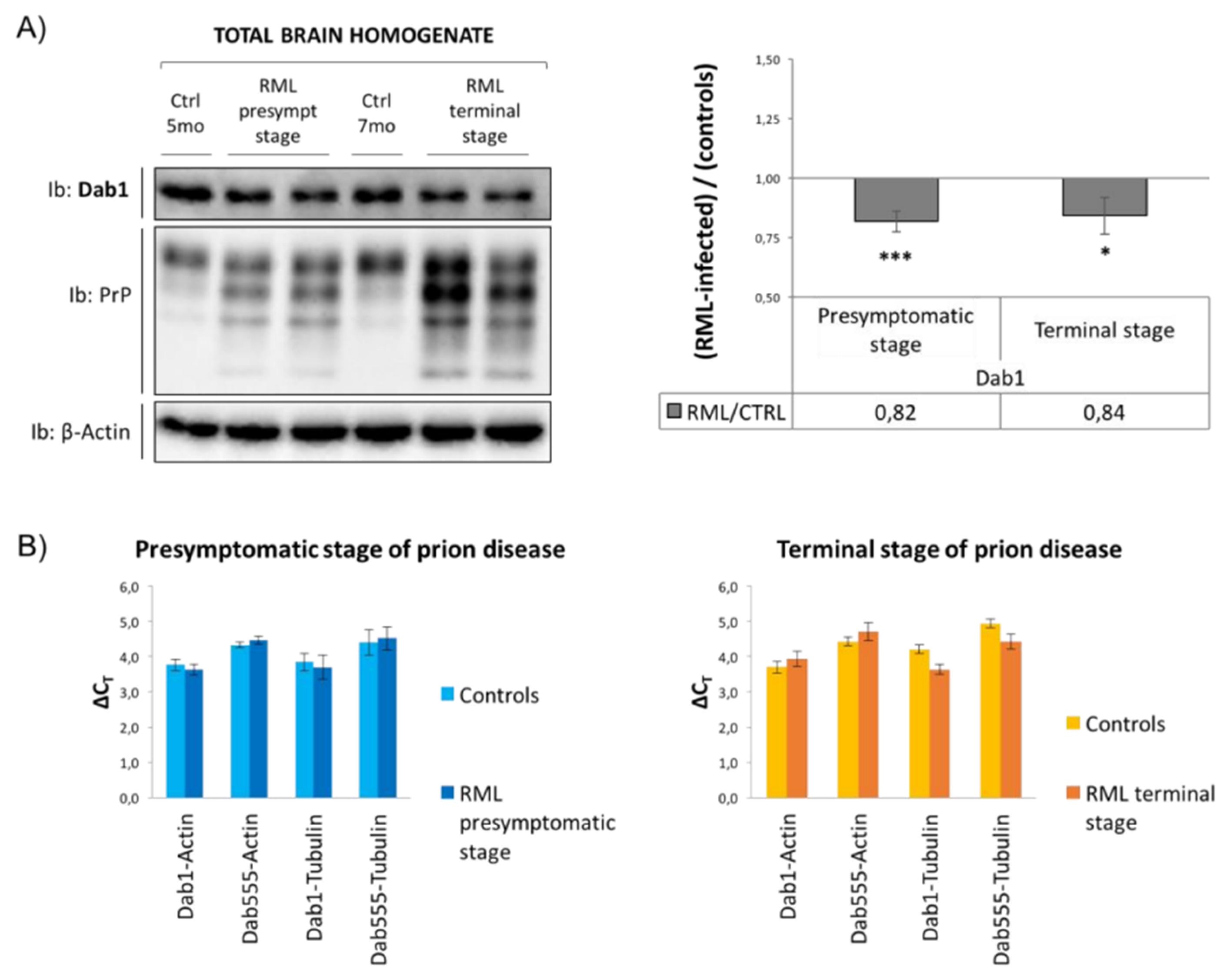
3.3. Analysis of Reelin/Dab1 Signaling Pathway in Prion-Infected Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Shattuck lecture--neurodegenerative diseases and prions. N. Engl. J. Med. 2001, 344, 1516–1526. [Google Scholar] [CrossRef]
- Soto, C.; Satani, N. The intricate mechanisms of neurodegeneration in prion diseases. Trends Mol. Med. 2011, 17, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular structure, biology, and genetics of prions. Adv. Virus Res. 1988, 35, 83–136. [Google Scholar]
- Cohen, F.E.; Pan, K.-M.; Huang, Z.; Baldwin, M.; Fletterick, R.J.; Prusiner, S.B. Structural clues to prion replication. Science 1994, 264, 530–531. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Prusiner, S.B. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990, 29, 5405–5412. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Didonna, A. Prion protein and its role in signal transduction. Cell. Mol. Biol. Lett. 2013, 18, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.Z.; Hernandez-Rapp, J.; Martin-Lannerée, S.; Launay, J.M.; Mouillet-Richard, S. PrP(C) signalling in neurons: From basics to clinical challenges. Biochimie 2014, 104, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef]
- Vassallo, N.; Herms, J.; Behrens, C.; Krebs, B.; Saeki, K.; Onodera, T.; Windl, O.; Kretzschmar, H.A. Activation of phosphatidylinositol 3-kinase by cellular prion protein and its role in cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 75–82. [Google Scholar] [CrossRef]
- Weise, J.; Sandau, R.; Schwarting, S.; Crome, O.; Wrede, A.; Schulz-Schaeffer, W.; Zerr, I.; BähR, M. Deletion of cellular prion protein results in reduced Akt activation, enhanced postischemic caspase-3 activation, and exacerbation of ischemic brain injury. Stroke 2006, 37, 1296–1300. [Google Scholar] [CrossRef]
- Hernandez-Rapp, J.; Martin-Lannerée, S.; Hirsch, T.Z.; Pradines, E.; Alleaume-Butaux, A.; Schneider, B.; Baudry, A.; Launay, J.-M.; Mouillet-Richard, S. A PrP(C)-caveolin-Lyn complex negatively controls neuronal GSK3beta and serotonin 1B receptor. Sci. Rep. 2014, 4, 4881. [Google Scholar] [CrossRef]
- Herz, J.; Chen, Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat. Rev. Neurosci. 2006, 7, 850–859. [Google Scholar] [CrossRef]
- Joly-Amado, A.; Kulkarni, N.; Nash, K.R. Reelin Signaling in Neurodevelopmental Disorders and Neurodegenerative Diseases. Brain Sci. 2023, 13, 1479. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Homayouni, R.; Keshvara, L.; Rice, D.S.; Sheldon, M.; Curran, T. Reelin is a ligand for lipoprotein receptors. Neuron 1999, 24, 471–479. [Google Scholar] [CrossRef]
- Hiesberger, T.; Trommsdorff, M.; Howell, B.W.; Goffinet, A.; Mumby, M.C.; Cooper, J.A.; Herz, J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron 1999, 24, 481–489. [Google Scholar] [CrossRef]
- Bock, H.H.; Herz, J. Reelin activates SRC family tyrosine kinases in neurons. Curr. Biol. 2003, 13, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, L.; Ballif, B.A.; Förster, E.; Cooper, J.A. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr. Biol. 2003, 13, 9–17. [Google Scholar] [CrossRef]
- Keshvara, L.; Benhayon, D.; Magdaleno, S.; Curran, T. Identification of reelin-induced sites of tyrosyl phosphorylation on disabled 1. J. Biol. Chem. 2001, 276, 16008–16014. [Google Scholar] [CrossRef] [PubMed]
- Bock, H.H.; Jossin, Y.; Liu, P.; Förster, E.; May, P.; Goffinet, A.M.; Herz, J. Phosphatidylinositol 3-kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J. Biol. Chem. 2003, 278, 38772–38779. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Goffinet, A.M. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol. Cell. Biol. 2007, 27, 7113–7124. [Google Scholar] [CrossRef]
- Beffert, U.; Morfini, G.; Bock, H.H.; Reyna, H.; Brady, S.T.; Herz, J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J. Biol. Chem. 2002, 277, 49958–49964. [Google Scholar] [CrossRef]
- Bock, H.H.; May, P. Canonical and Non-canonical Reelin Signaling. Front. Cell. Neurosci. 2016, 10, 166. [Google Scholar] [CrossRef]
- Ballif, B.A.; Arnaud, L.; Arthur, W.T.; Guris, D.; Imamoto, A.; Cooper, J.A. Activation of a Dab1/CrkL/C3G/Rap1 pathway in Reelin-stimulated neurons. Curr. Biol. 2004, 14, 606–610. [Google Scholar] [CrossRef]
- Mata, A.; Urrea, L.; Vilches, S.; Llorens, F.; Thüne, K.; Espinosa, J.-C.; Andréoletti, O.; Sevillano, A.M.; Torres, J.M.; Requena, J.R.; et al. Reelin Expression in Creutzfeldt-Jakob Disease and Experimental Models of Transmissible Spongiform Encephalopathies. Mol. Neurobiol. 2017, 54, 6412–6425. [Google Scholar] [CrossRef]
- Lidón, L.; Urrea, L.; Llorens, F.; Gil, V.; Alvarez, I.; Diez-Fairen, M.; Aguilar, M.; Pastor, P.; Zerr, I.; Alcolea, D.; et al. Disease-Specific Changes in Reelin Protein and mRNA in Neurodegenerative Diseases. Cells 2020, 9, 1252. [Google Scholar] [CrossRef] [PubMed]
- Herrick, T.M.; Cooper, J.A. A hypomorphic allele of dab1 reveals regional differences in reelin-Dab1 signaling during brain development. Development 2002, 129, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Leemhuis, J.; Bouché, E.; Frotscher, M.; Henle, F.; Hein, L.; Herz, J.; Meyer, D.K.; Pichler, M.; Roth, G.; Schwan, C.; et al. Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. J. Neurosci. 2010, 30, 14759–14772. [Google Scholar] [CrossRef] [PubMed]
- Peretz, D.; Williamson, R.A.; Kaneko, K.; Vergara, J.; Leclerc, E.; Schmitt-Ulms, G.; Mehlhorn, I.R.; Legname, G.; Wormald, M.R.; Rudd, P.M.; et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 2001, 412, 739–743. [Google Scholar] [CrossRef]
- Petsch, B.; Müller-Schiffmann, A.; Lehle, A.; Zirdum, E.; Prikulis, I.; Kuhn, F.; Raeber, A.J.; Ironside, J.W.; Korth, C.; Stitz, L. Biological effects and use of PrPSc- and PrP-specific antibodies generated by immunization with purified full-length native mouse prions. J. Virol. 2011, 85, 4538–4546. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bock, H.H.; Jossin, Y.; May, P.; Bergner, O.; Herz, J. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J. Biol. Chem. 2004, 279, 33471–33479. [Google Scholar] [CrossRef]
- Grison, A.; Zucchelli, S.; Urzì, A.; Zamparo, I.; Lazarevic, D.; Pascarella, G.; Roncaglia, P.; Giorgetti, A.; Garcia-Esparcia, P.; Vlachouli, C.; et al. Mesencephalic dopaminergic neurons express a repertoire of olfactory receptors and respond to odorant-like molecules. BMC Genom. 2014, 15, 729. [Google Scholar] [CrossRef]
- Simonetti, M.; Giniatullin, R.; Fabbretti, E. Mechanisms mediating the enhanced gene transcription of P2 × 3 receptor by calcitonin gene-related peptide in trigeminal sensory neurons. J. Biol. Chem. 2008, 283, 18743–18752. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef]
- Barbisin, M.; Vanni, S.; Schmädicke, A.-C.; Montag, J.; Motzkus, D.; Opitz, L.; Salinas-Riester, G.; Legname, G. Gene expression profiling of brains from bovine spongiform encephalopathy (BSE)-infected cynomolgus macaques. BMC Genom. 2014, 15, 434. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Kaneda, T.; Muto, A.; Yoshida, T. Positive transcriptional regulation of the human micro opioid receptor gene by poly(ADP-ribose) polymerase-1 and increase of its DNA binding affinity based on polymorphism of G-172 → T. J. Biol. Chem. 2009, 284, 20175–20183. [Google Scholar] [CrossRef]
- Kohno, S.; Kohno, T.; Nakano, Y.; Suzuki, K.; Ishii, M.; Tagami, H.; Baba, A.; Hattori, M. Mechanism and significance of specific proteolytic cleavage of Reelin. Biochem. Biophys. Res. Commun. 2009, 380, 93–97. [Google Scholar] [CrossRef]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Jossin, Y. Reelin Functions, Mechanisms of Action and Signaling Pathways During Brain Development and Maturation. Biomolecules 2020, 10, 964. [Google Scholar] [CrossRef] [PubMed]
- Alexander, A.; Herz, J.; Calvier, L. Reelin through the years: From brain development to inflammation. Cell Rep. 2023, 42, 112669. [Google Scholar] [CrossRef]
- Hoe, H.-S.; Tran, T.S.; Matsuoka, Y.; Howell, B.W.; Rebeck, G.W. DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J. Biol. Chem. 2006, 281, 35176–35185. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.E.; Zamora, D.; Horowitz, M.S.; Jahanipour, J.; Calzada, E.; Li, X.; Keyes, G.S.; Murray, H.C.; Curtis, M.A.; Faull, R.M.; et al. ApoER2-Dab1 disruption as the origin of pTau-associated neurodegeneration in sporadic Alzheimer’s disease. Acta Neuropathol. Commun. 2023, 11, 197. [Google Scholar] [CrossRef]
- Gavín, R.; Ferrer, I.; del Río, J.A. Involvement of Dab1 in APP processing and beta-amyloid deposition in sporadic Creutzfeldt-Jakob patients. Neurobiol. Dis. 2010, 37, 324–329. [Google Scholar] [CrossRef]
- Benvegnu, S.; Poggiolini, I.; Legname, G. Neurodevelopmental expression and localization of the cellular prion protein in the central nervous system of the mouse. J. Comp. Neurol. 2010, 518, 1879–1891. [Google Scholar] [CrossRef]
- Alcantara, S.; Ruiz, M.; D’Arcangelo, G.; Ezan, F.; de Lecea, L.; Curran, T.; Sotelo, C.; Soriano, E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J. Neurosci. 1998, 18, 7779–7799. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Allen, N.S.; Simo, S.; Cooper, J.A. Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 2007, 21, 2717–2730. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, M.; Rice, D.S.; D’ARcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.S.; Sheldon, M.; D’aRcangelo, G.; Nakajima, K.; Goldowitz, D.; Curran, T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 1998, 125, 3719–3729. [Google Scholar] [CrossRef]
- Trommsdorff, M.; Gotthardt, M.; Hiesberger, T.; Shelton, J.; Stockinger, W.; Nimpf, J.; Hammer, R.E.; Richardson, J.A.; Herz, J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 1999, 97, 689–701. [Google Scholar] [CrossRef]
- Kuo, G.; Arnaud, L.; Kronstad-O’BRien, P.; Cooper, J.A. Absence of Fyn and Src causes a reeler-like phenotype. J. Neurosci. 2005, 25, 8578–8586. [Google Scholar] [CrossRef]
- Arnaud, L.; Ballif, B.A.; Cooper, J.A. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol. Cell. Biol. 2003, 23, 9293–9302. [Google Scholar] [CrossRef]
- Kerjan, G.; Gleeson, J.G. A missed exit: Reelin sets in motion Dab1 polyubiquitination to put the break on neuronal migration. Genes Dev. 2007, 21, 2850–2854. [Google Scholar] [CrossRef]
- Lussier, A.L.; Weeber, E.J.; Rebeck, G.W. Reelin Proteolysis Affects Signaling Related to Normal Synapse Function and Neurodegeneration. Front. Cell. Neurosci. 2016, 10, 75. [Google Scholar] [CrossRef]
- Sáez-Valero, J.; Costell, M.; Sjögren, M.; Andreasen, N.; Blennow, K.; Luque, J.M. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer’s disease. J. Neurosci. Res. 2003, 72, 132–136. [Google Scholar] [CrossRef]
- Botella-López, A.; Burgaya, F.; Gavín, R.; García-Ayllón, M.S.; Gómez-Tortosa, E.; Peña-Casanova, J.; Ureña, J.M.; Del Río, J.A.; Blesa, R.; Soriano, E.; et al. Reelin expression and glycosylation patterns are altered in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5573–5578. [Google Scholar] [CrossRef]
- Todaro, L.; Christiansen, S.; Varela, M.; Campodónico, P.; Pallotta, M.G.; Lastiri, J.; de Lustig, E.S.; Joffé, E.B.d.K.; Puricelli, L. Alteration of serum and tumoral neural cell adhesion molecule (NCAM) isoforms in patients with brain tumors. J. Neuro-Oncol. 2007, 83, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Völker, H.-U.; Engert, S.; Cramer, A.; Schmidt, M.; Kämmerer, U.; Müller-Hermelink, H.-K.; Gattenlöhner, S. Expression of CD56 isoforms in primary and relapsed adult granulosa cell tumors of the ovary. Diagn. Pathol. 2008, 3, 29. [Google Scholar] [CrossRef]
- Marković-Lipkovski, J.; Životić, M.; Müller, C.A.; Tampe, B.; Ćirović, S.; Vještica, J.; Tomanović, N.; Zeisberg, M.; Müller, G.A.; Singh, S.R. Variable Expression of Neural Cell Adhesion Molecule Isoforms in Renal Tissue: Possible Role in Incipient Renal Fibrosis. PLoS ONE 2015, 10, e0137028. [Google Scholar] [CrossRef] [PubMed]
- Tur, M.K.; Etschmann, B.; Benz, A.; Leich, E.; Waller, C.; Schuh, K.; Rosenwald, A.; Ertl, G.; Kienitz, A.; Haaf, A.T.; et al. The 140-kD isoform of CD56 (NCAM1) directs the molecular pathogenesis of ischemic cardiomyopathy. Am. J. Pathol. 2013, 182, 1205–1218. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, A.; Burwinkel, M.; Riemer, C.; Schultz, J.; Baier, M. Unchanged scrapie pathology in brain tissue of tyrosine kinase Fyn-deficient mice. Neurodegener. Dis. 2004, 1, 266–268. [Google Scholar] [CrossRef]
- Nixon, R.R. Prion-associated increases in Src-family kinases. J. Biol. Chem. 2005, 280, 2455–2462. [Google Scholar] [CrossRef]
- Gyllberg, H.; Löfgren, K.; Lindegren, H.; Bedecs, K. Increased Src kinase level results in increased protein tyrosine phosphorylation in scrapie-infected neuronal cell lines. FEBS Lett. 2006, 580, 2603–2608. [Google Scholar] [CrossRef]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Antibody | Company/Reference | Dilution |
|---|---|---|---|
| Reelin | Mouse monoclonal G10 | Millipore | 1:1000 |
| Reelin | Mouse monoclonal 20E12 | Prof. Korth’s laboratory | IP |
| DISC1 | Mouse monoclonal 14F2 | Prof. Korth’s laboratory | IP |
| ApoER2 | Rabbit monoclonal [EPR3326] | Abcam | 1:1000 |
| VLDLR | Mouse monoclonal 6A6 | Millipore | 1:1000 |
| Dab1 | Rabbit monoclonal [EP2248Y] | Abcam | 1:1000 |
| Dab1 | Rabbit polyclonal H-103 | Santa Cruz Biotechnology | 1:500 |
| Phospho-Tyrosine | Mouse monoclonal 4G10 | Millipore | 1:1000 |
| Fyn | Rabbit monoclonal 04-343 | Millipore | 1:2000 |
| Fyn | Mouse monoclonal (FYN-59) | Santa Cruz Biotechnology | 1:1000 |
| Phospho-Src family (Tyr416) | Rabbit polyclonal 2101S | Cell Signaling Technologies | 1:1000 |
| NCAM | Rabbit polyclonal AB5032 | Chemicon International | 1:1000 |
| AKT | Rabbit polyclonal 9272 | Cell Signaling Technologies | 1:1000 |
| Prion protein | Humanized D18 antibody fragment (Fab) | [34] | 1:1000 |
| Prion protein | Mouse monoclonal W226 | [35] | 1:1000 |
| Target | Primer Name | Sequence (5′-3′) | Amplicon Size (bp) | Accession Number | Reference |
|---|---|---|---|---|---|
| Mouse Dab1 | MoDab1_fw MoDab1_rev | GCCAAGAAAGACTCCAGGAAGA GGACCCCTTCGCCTTTAAAC | 79 | NM_177259.4 NM_010014.3 | [37] |
| Mouse Dab1 | MoDab555_fw MoDab555_rev | TTATGATGTGCCAAAAAGTCAACCT AGTTCTAGTTGGGTCACAGCACTTAC | 51 | NM_177259.4 | [37] |
| Mouse β-Actin | MoActb_fw MoActb_rev | CACACCCGCCACCAGTTC CCCATTCCCACCATCACACC | 164 | NM_007393.5 | [38] |
| Mouse βIII-Tubulin | MoTubb3_fw MoTubb3_rev | CGCCTTTGGACACCTATTC TACTCCTCACGCACCTTG | 240 | NM_023279.2 | [39] |
| Human Dab1 | HuDab1_fw HuDab1_rev | CACCGGGCCTTTGGATATGT GAATAACAGGTTCAGCCGCC | 88 | NM_021080.3 | Designed |
| Human β-Actin | HuActb_fw HuActb_rev | AGAGCTACGAGCTGCCTGAC AGCACTGTGTTGGCGTACAG | 184 | NM_001101.3 | [40] |
| Human GAPDH | HuGAPDH_fw HuGAPDH_rev | CCTGCACCACCAACTGCTTA TCTTCTGGGTGGCAGTGATG | 108 | NM_001289746.1 | Modified from [41] |
| Human RpL19 | HuRpL19_fw HuRpL19_rev | CTAGTGTCCTCCGCTGTGG AAGGTGTTTTTCCGGCATC | 169 | NM_000981.3 | [42] |
| IP Target | Probed For | Expectation | Result | Controls Used |
|---|---|---|---|---|
| PrPC (W226) | Reelin | Test | ✗ | PFHM, anti-DISC1 |
| PrPC (W226) | ApoER2 | Test | ✗ | PFHM, anti-DISC1 |
| Reelin (20E12) | PrPC | Test | ✗ | Normal mouse IgG |
| ApoER2 (EPR3326) | PrPC | Test | ✗ | Normal rabbit IgG |
| Reelin (20E12) | ApoER2 | Positive control | ✓ | Normal mouse IgG |
| ApoER2 (EPR3326) | Reelin | Positive control | ✓ | Normal rabbit IgG |
| DISC1 (14F2) | Reelin, ApoER2, and PrPC | Negative control | ✗ | N/A |
| PFHM (no Ab) | Reelin, ApoER2, and PrPC | Negative control | ✗ | N/A |
| Normal Mouse IgG | Reelin, ApoER2, and PrPC | Negative control | ✗ | N/A |
| Normal Mouse IgG | Reelin, ApoER2, and PrPC | Negative control | ✗ | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rolle, I.G.; Burato, A.; Bacınoğlu, M.B.; Moda, F.; Legname, G. The Role of Prion Protein in Reelin/Dab1 Signaling: Implications for Neurodegeneration. Viruses 2025, 17, 928. https://doi.org/10.3390/v17070928
Rolle IG, Burato A, Bacınoğlu MB, Moda F, Legname G. The Role of Prion Protein in Reelin/Dab1 Signaling: Implications for Neurodegeneration. Viruses. 2025; 17(7):928. https://doi.org/10.3390/v17070928
Chicago/Turabian StyleRolle, Irene Giulia, Anna Burato, Merve Begüm Bacınoğlu, Fabio Moda, and Giuseppe Legname. 2025. "The Role of Prion Protein in Reelin/Dab1 Signaling: Implications for Neurodegeneration" Viruses 17, no. 7: 928. https://doi.org/10.3390/v17070928
APA StyleRolle, I. G., Burato, A., Bacınoğlu, M. B., Moda, F., & Legname, G. (2025). The Role of Prion Protein in Reelin/Dab1 Signaling: Implications for Neurodegeneration. Viruses, 17(7), 928. https://doi.org/10.3390/v17070928