



Viruses 2024, 16(1), 69; https://doi.org/10.3390/v16010069 - 30 Dec 2023
Cited by 2 | Viewed by 2253
Abstract
►
Show Figures
Throughout the COVID-19 pandemic, an unprecedented level of clinical nasal swab data from around the globe has been collected and shared. Positive tests have consistently revealed viral titers spanning six orders of magnitude! An open question is whether such extreme population heterogeneity is
[...] Read more.
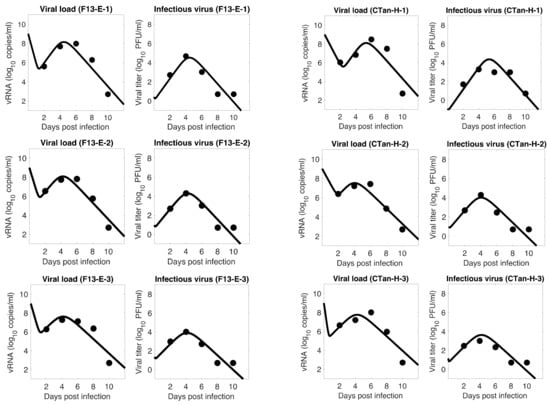
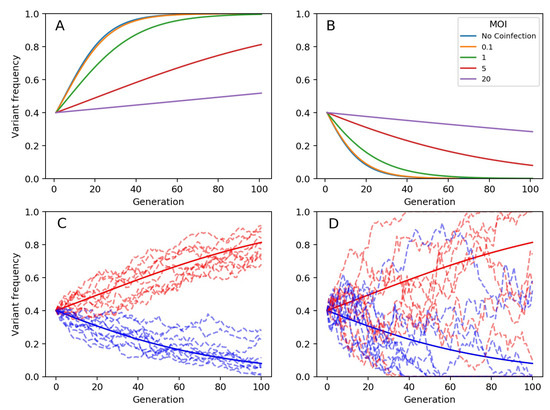
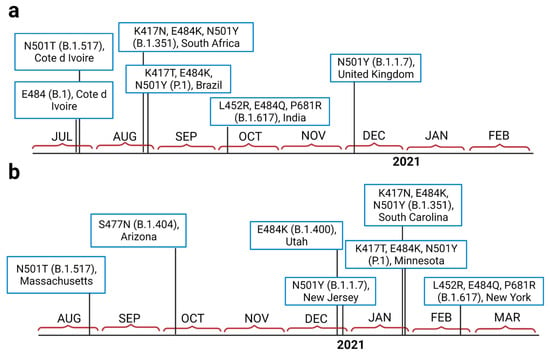
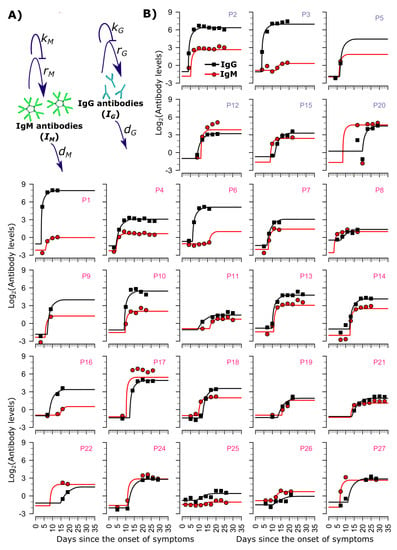
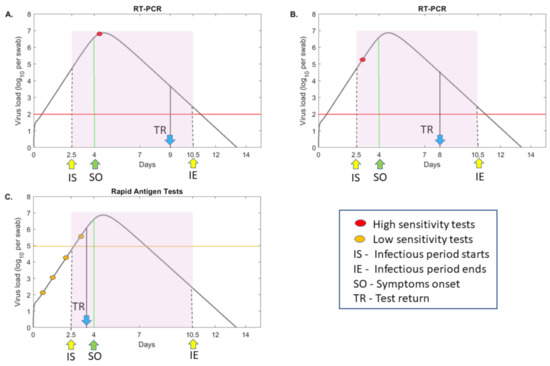
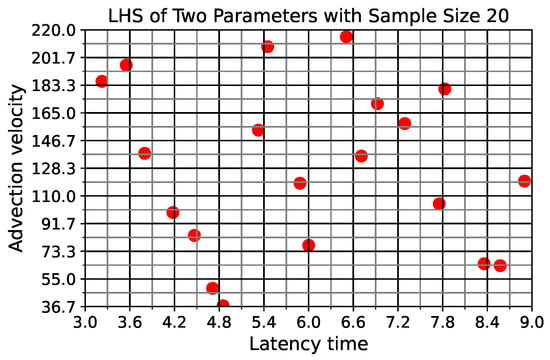
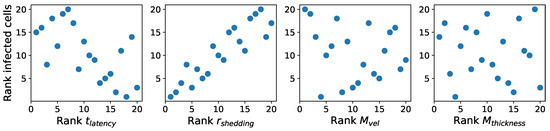
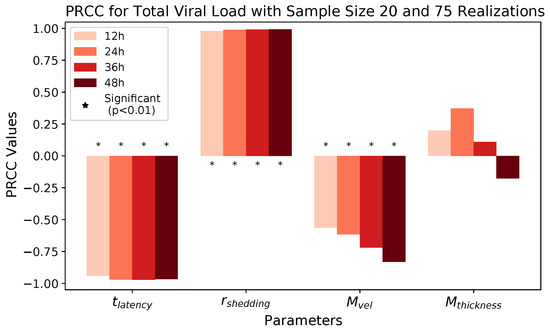
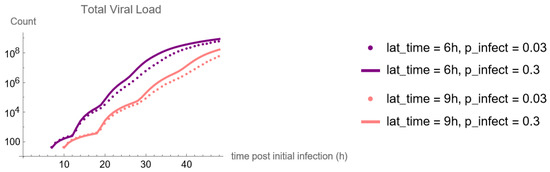
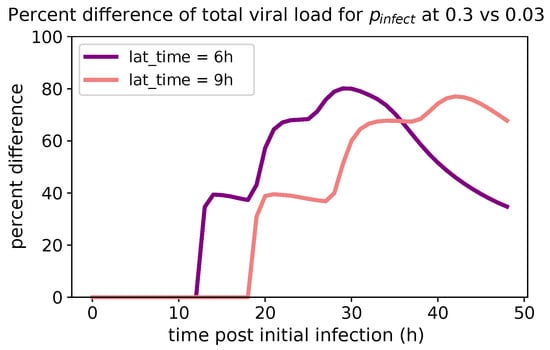
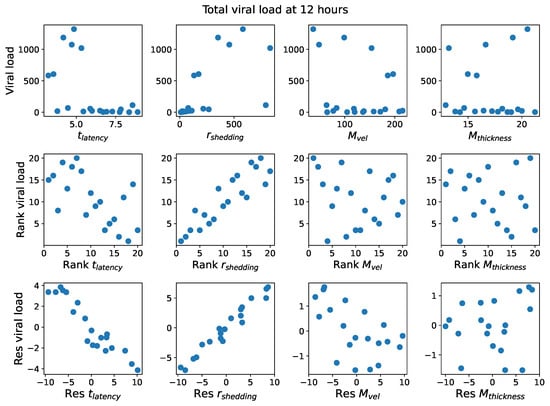
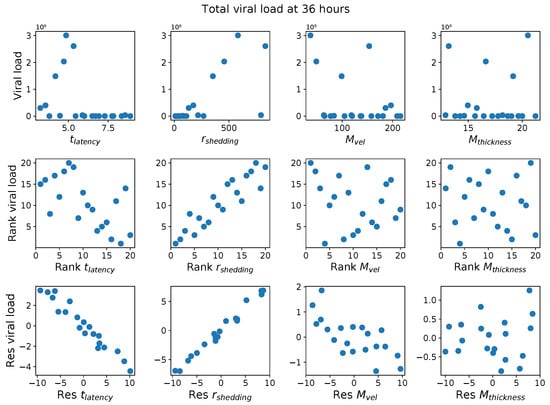
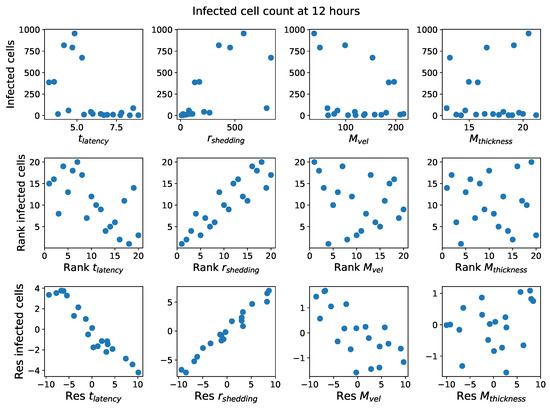
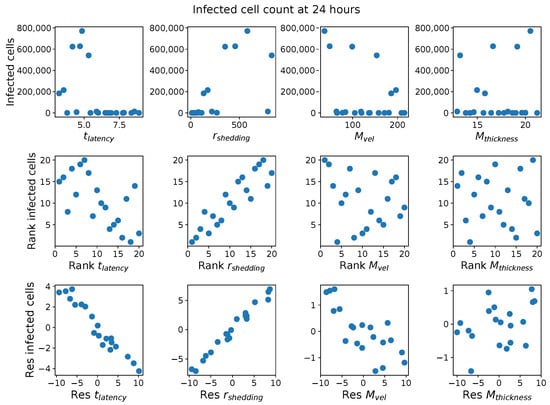
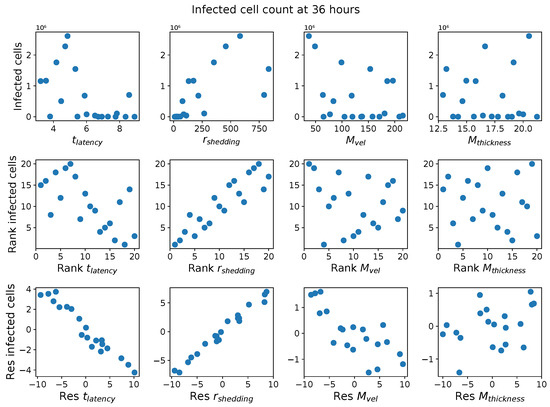
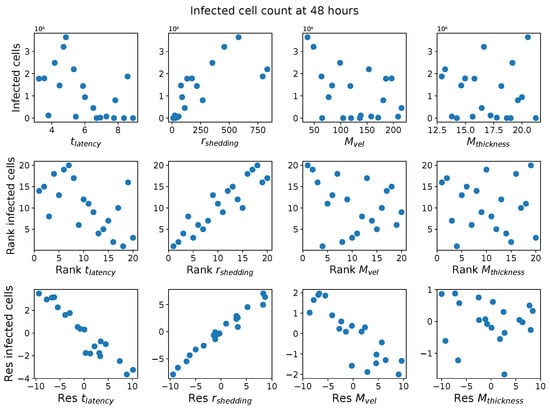
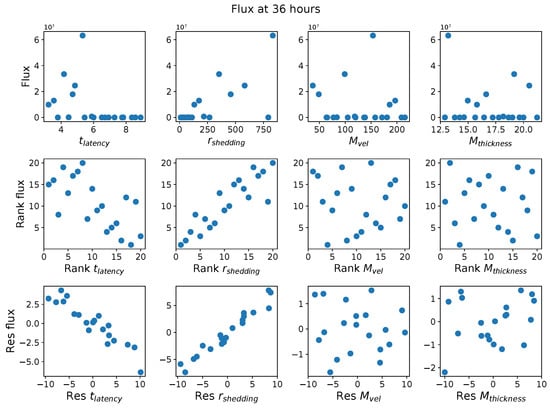
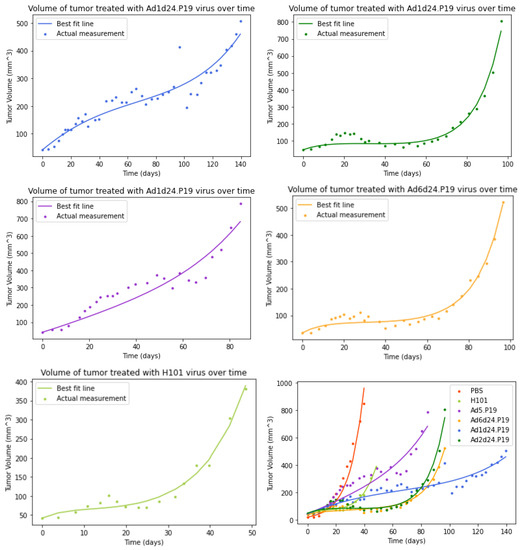
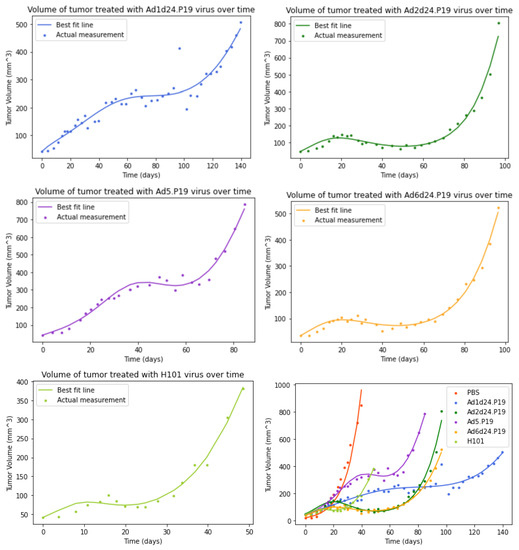
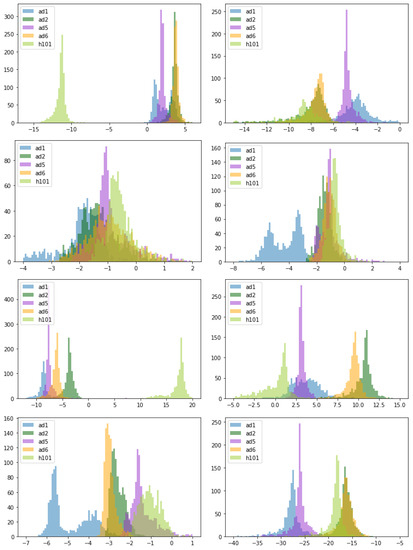
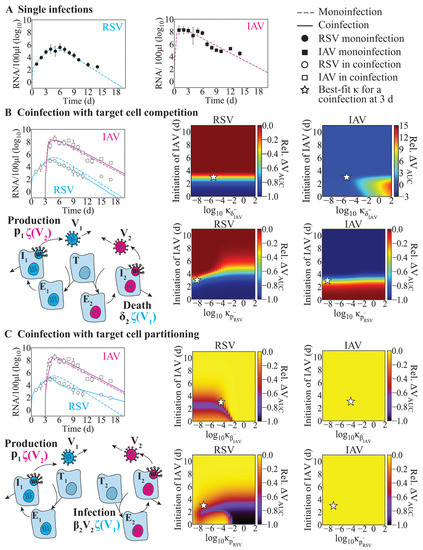
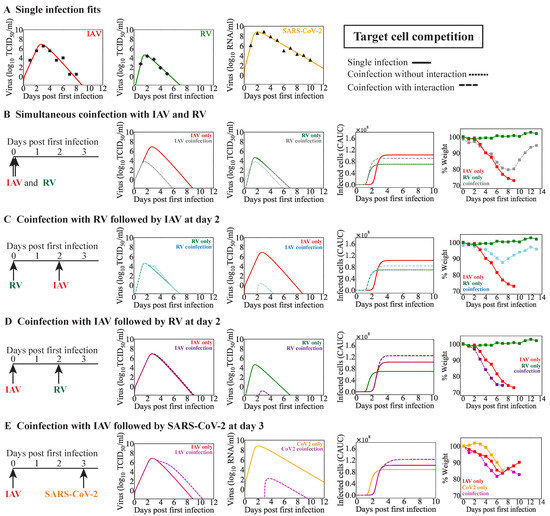
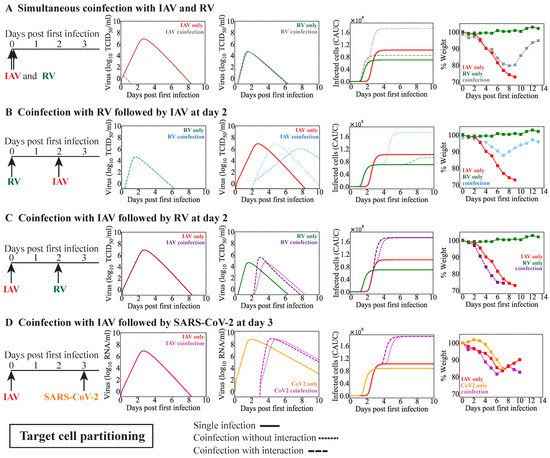
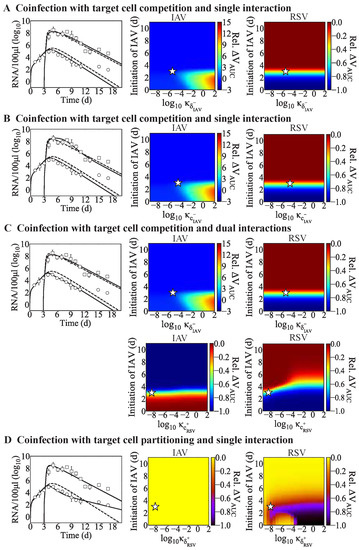
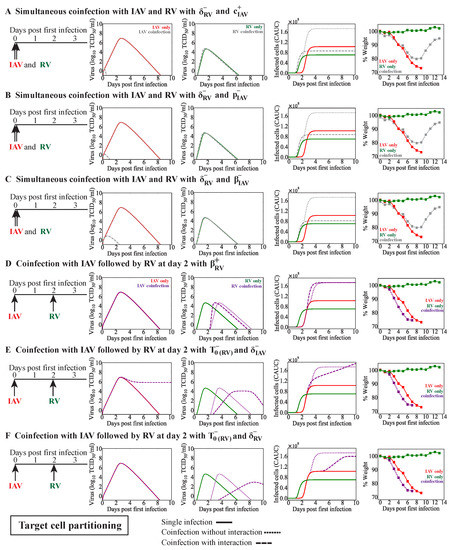
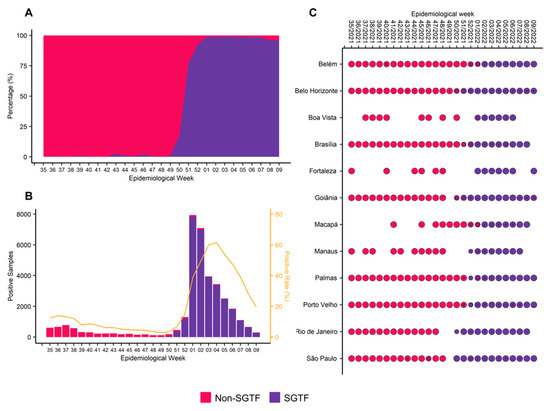
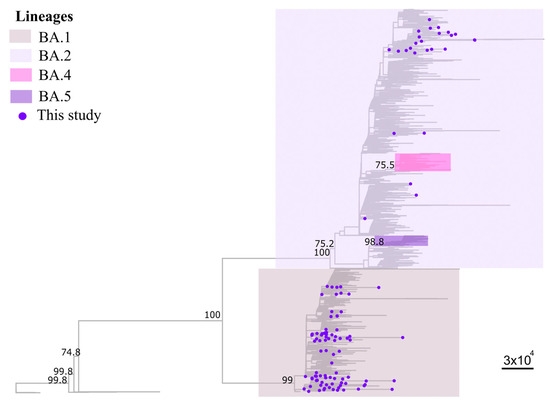
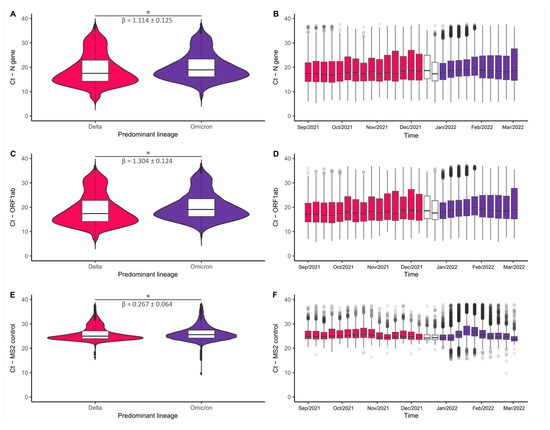
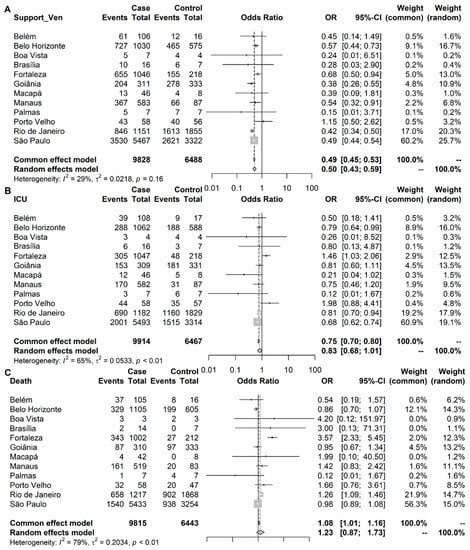
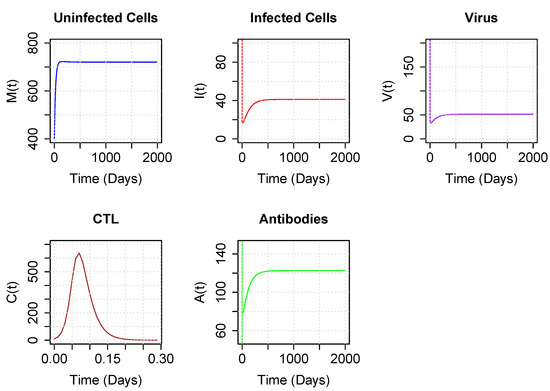
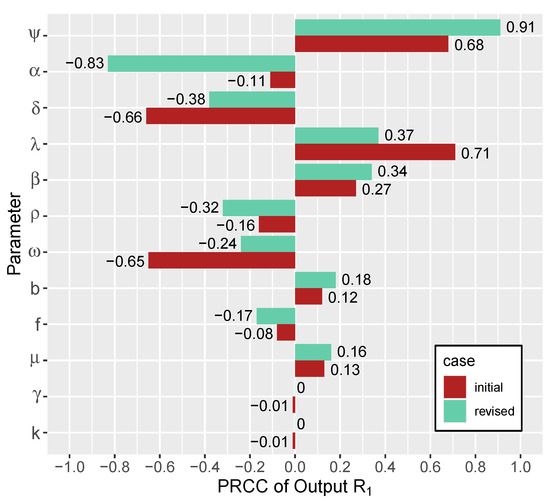
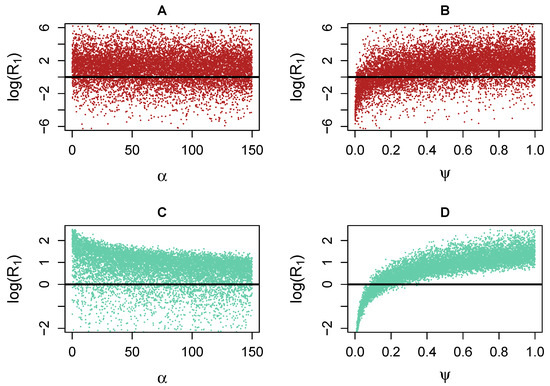
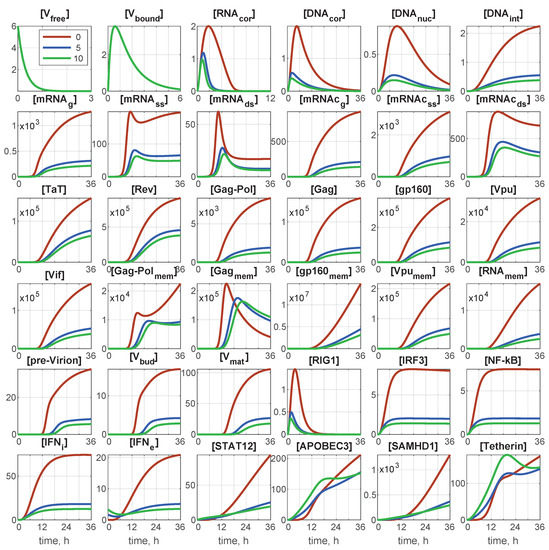
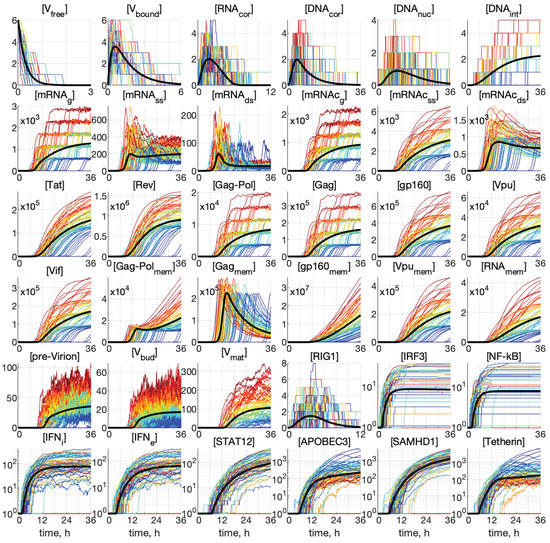
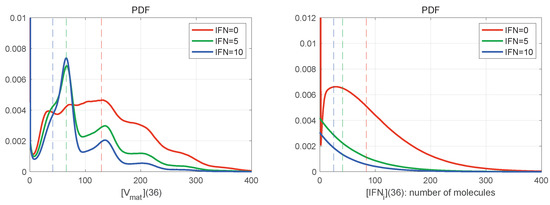
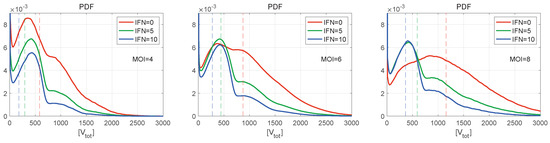
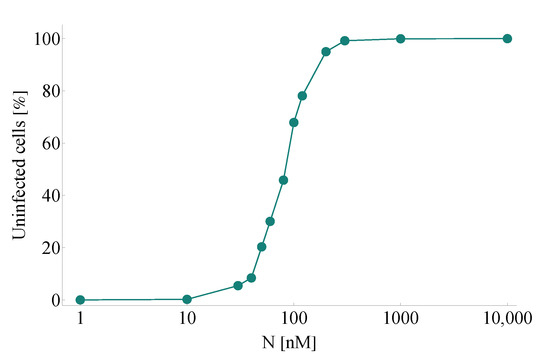
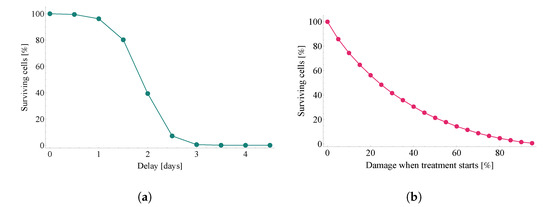
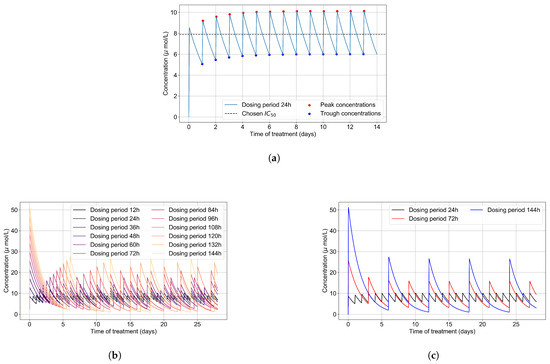
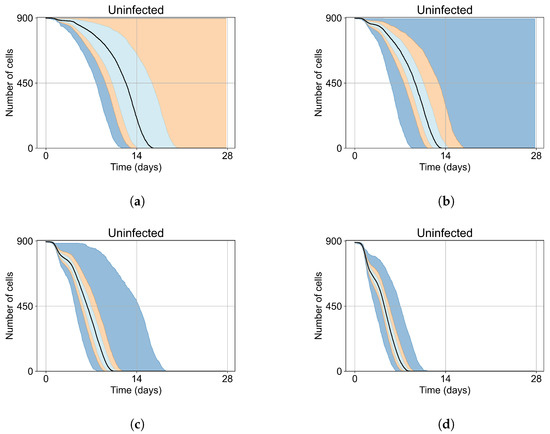
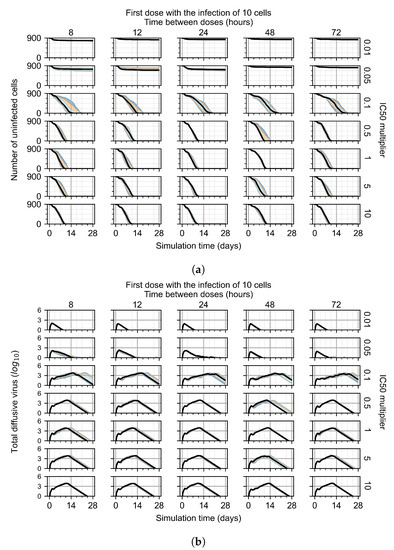
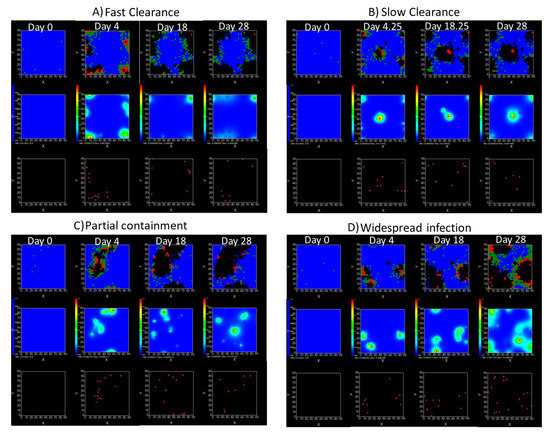
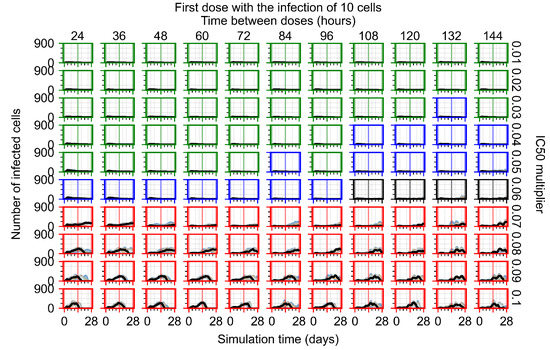
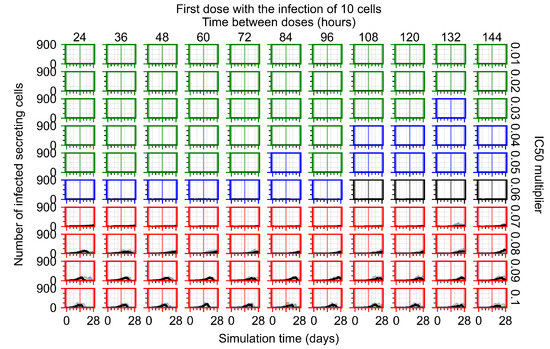
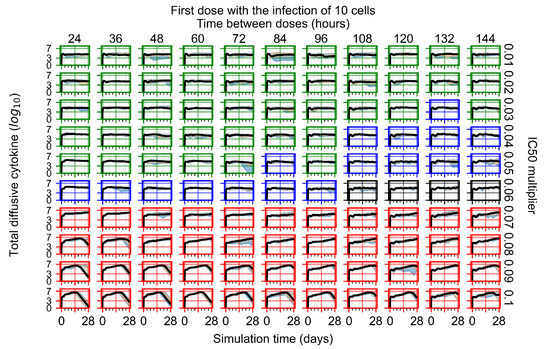
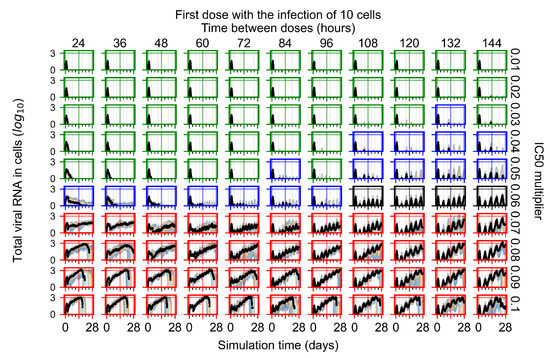
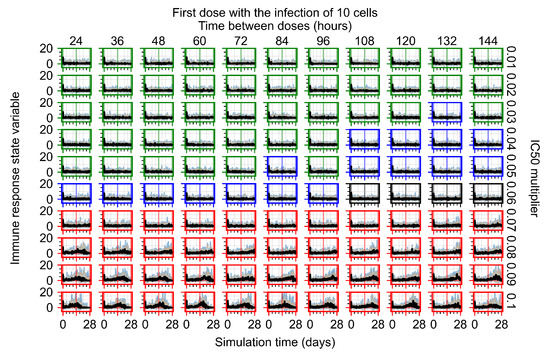
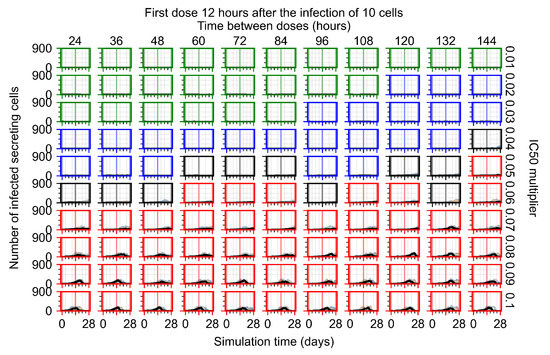
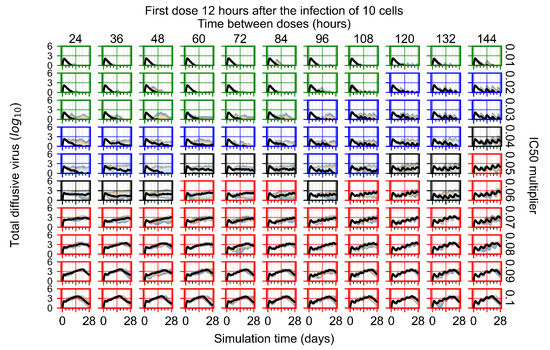
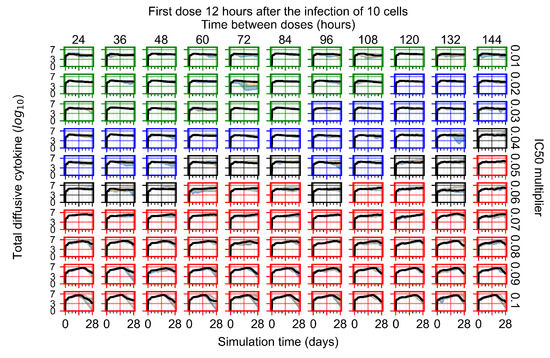
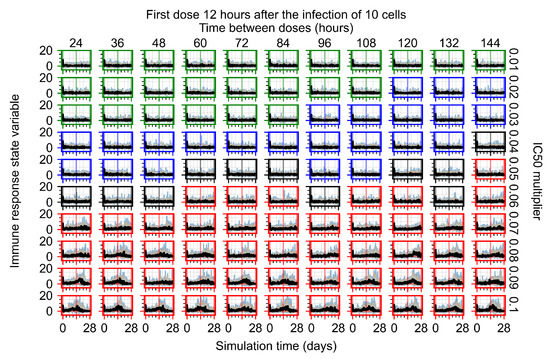
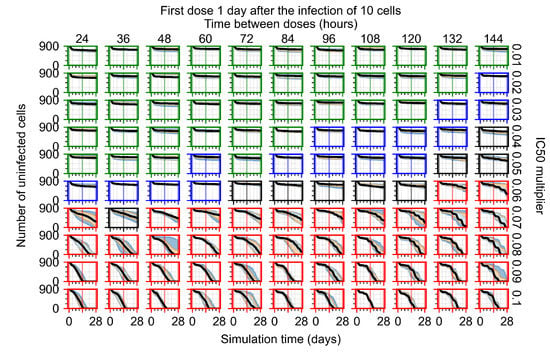
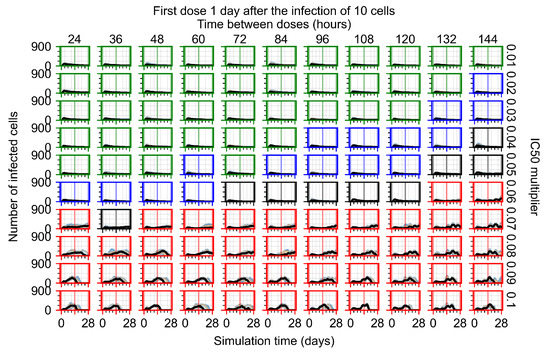
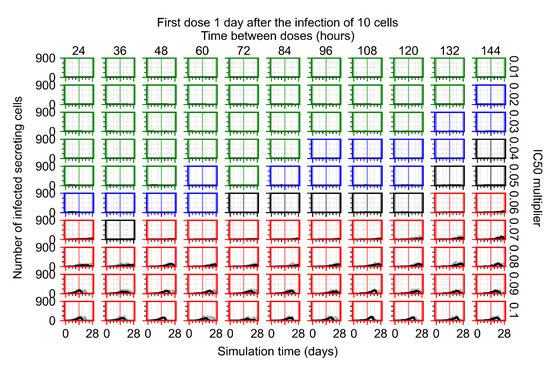
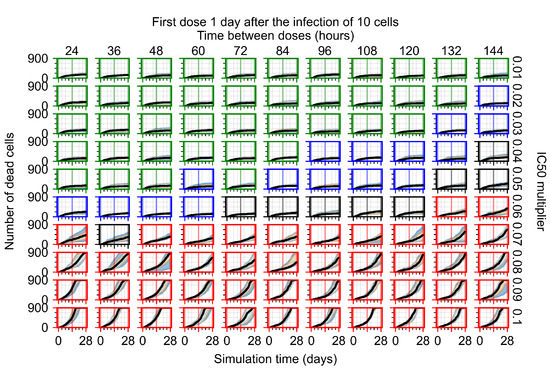
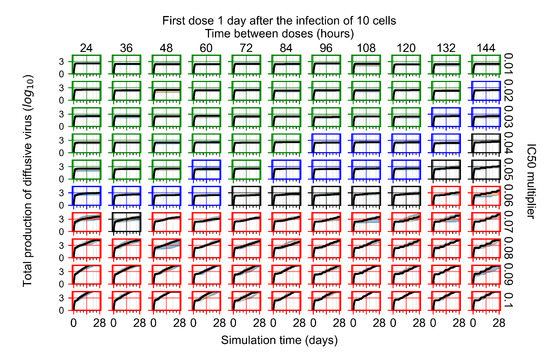
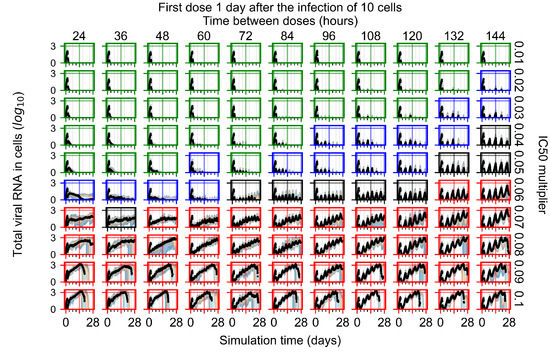
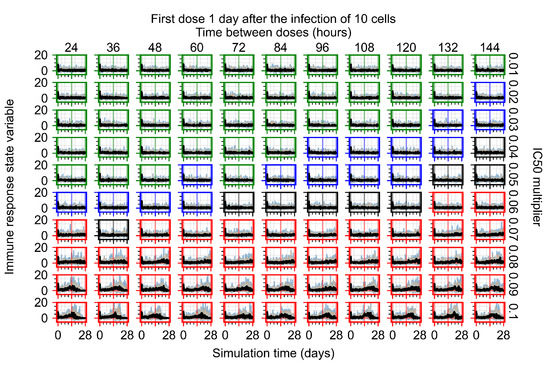
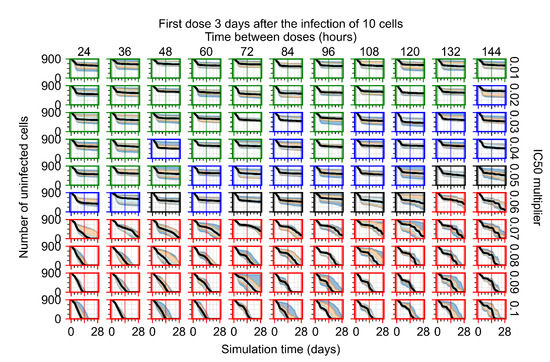
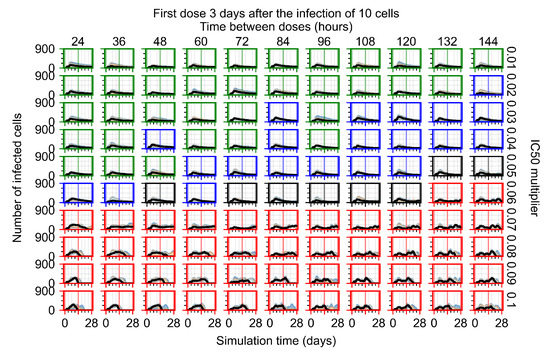
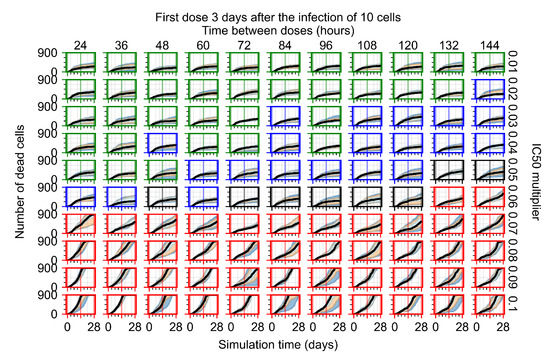
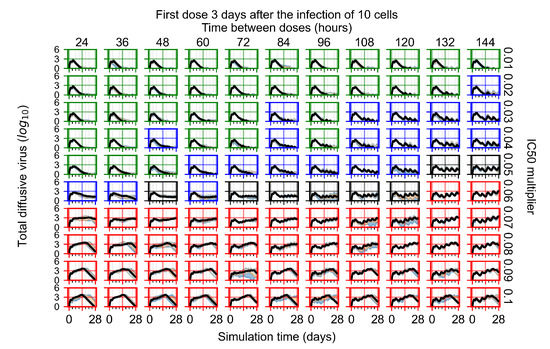
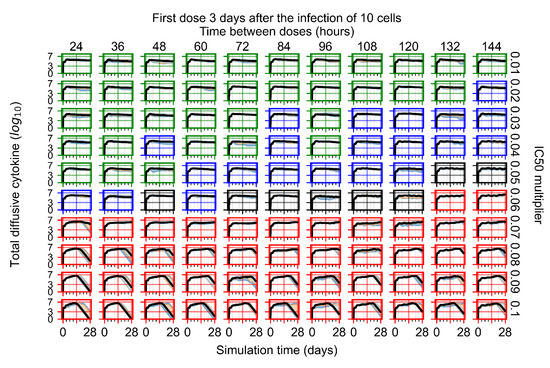
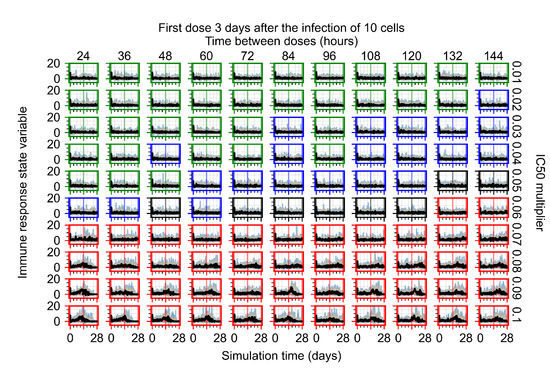
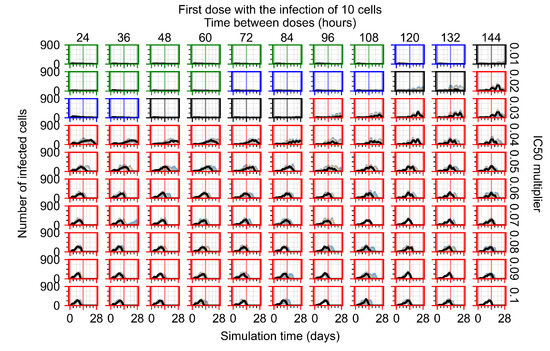
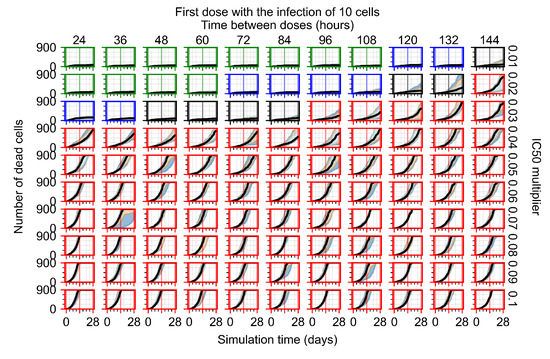
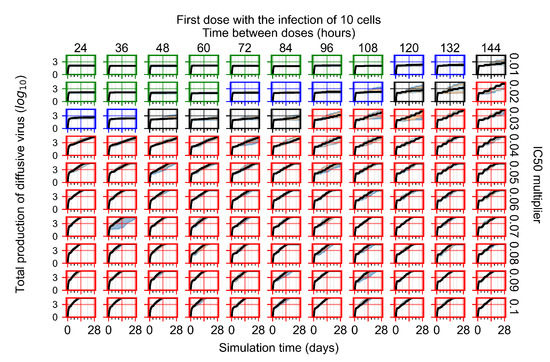
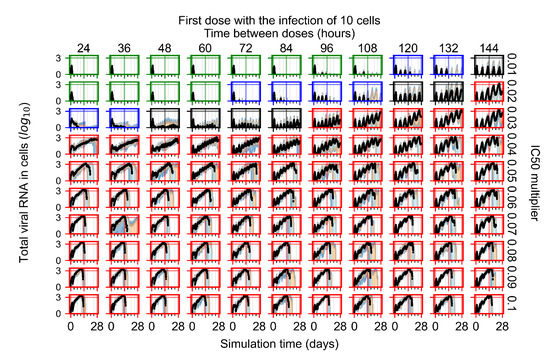
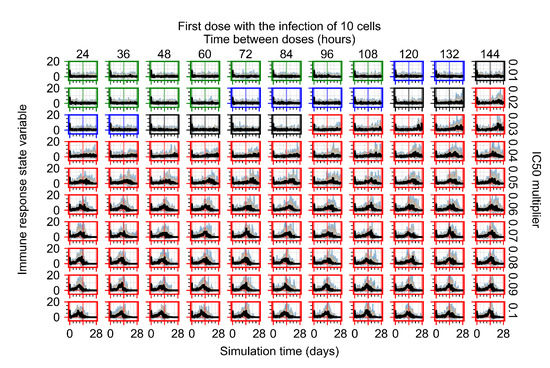
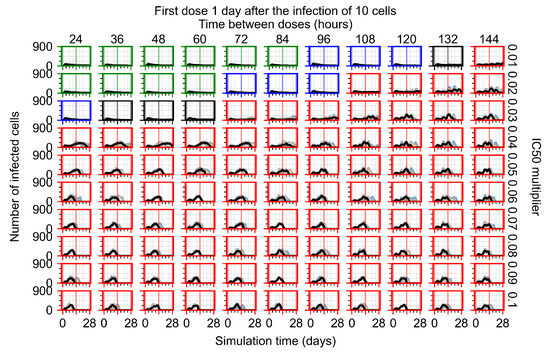
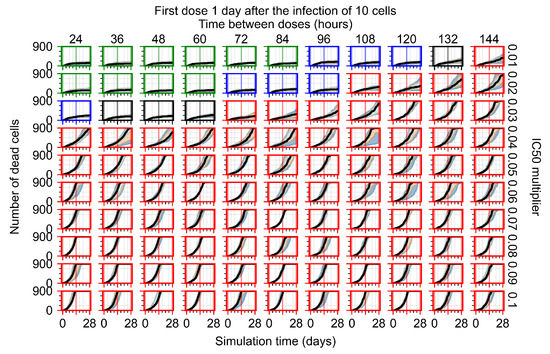
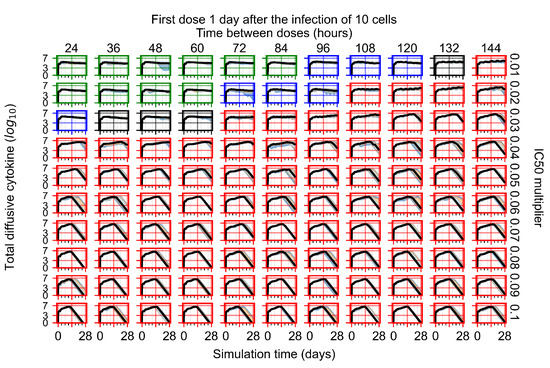
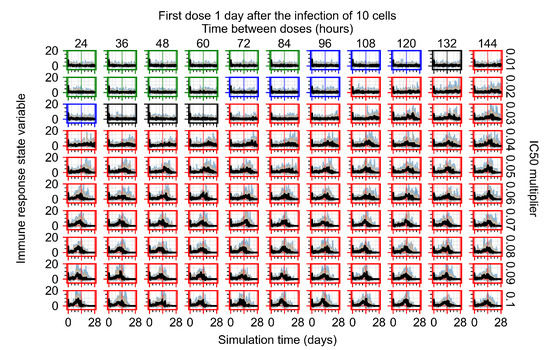
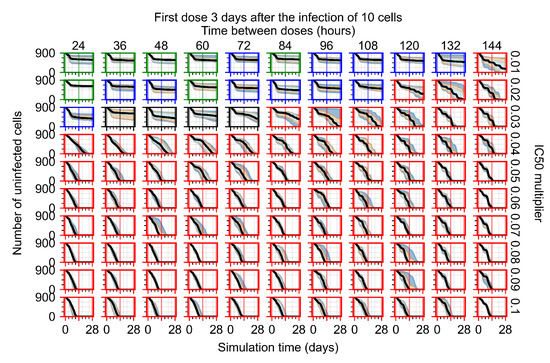
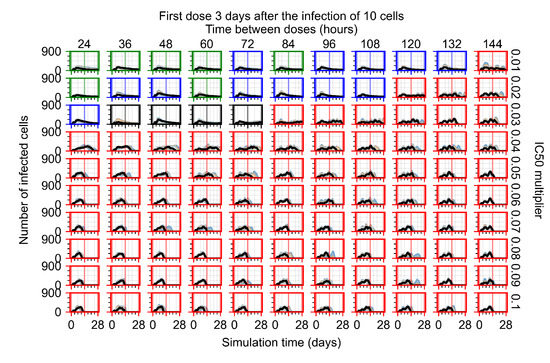
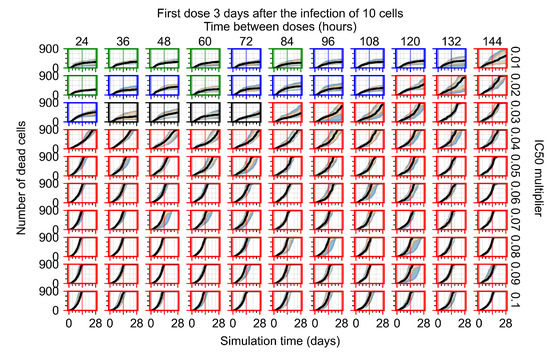
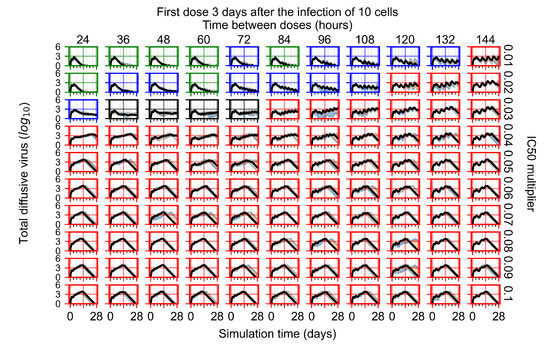
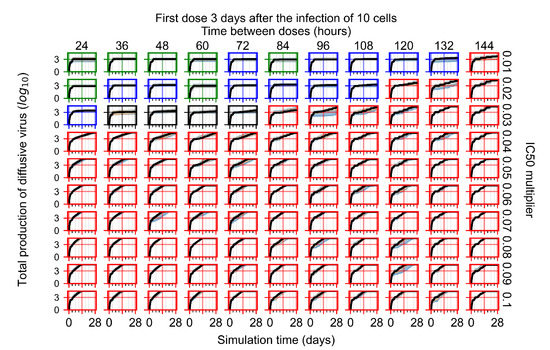
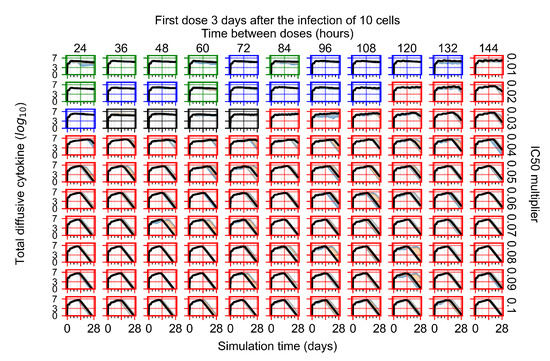
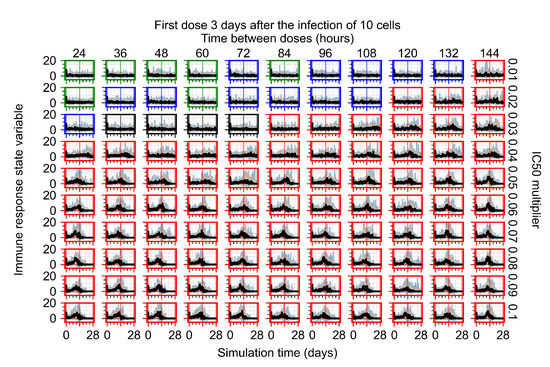
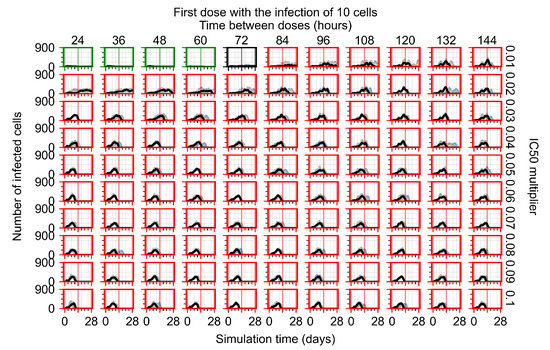
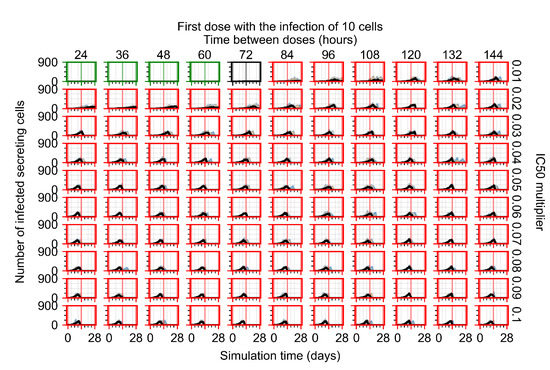
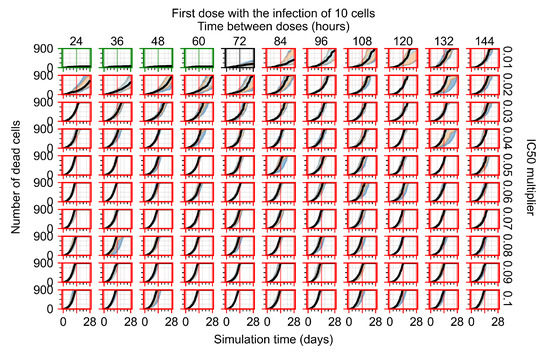
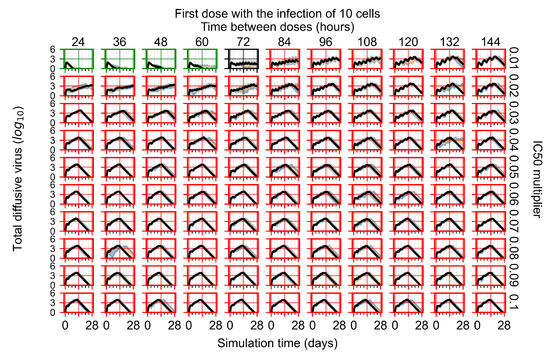
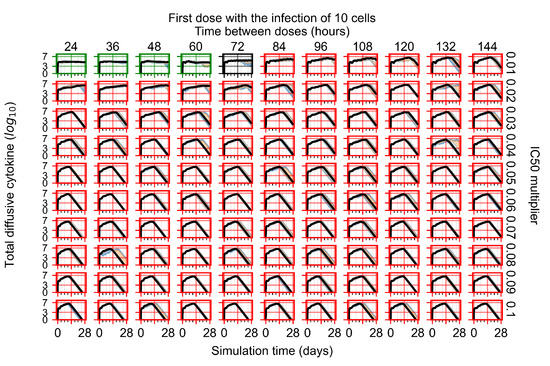
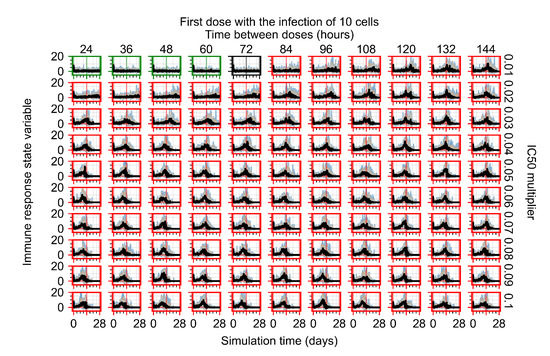
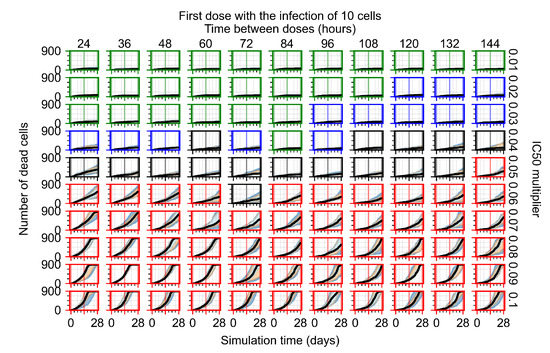
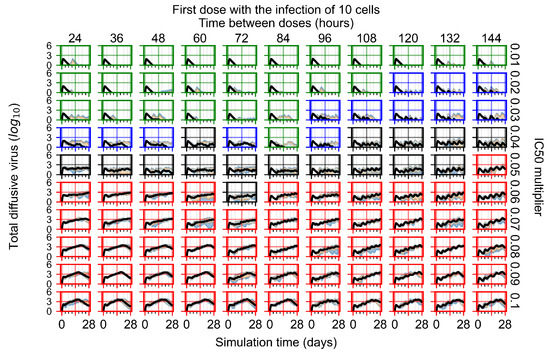
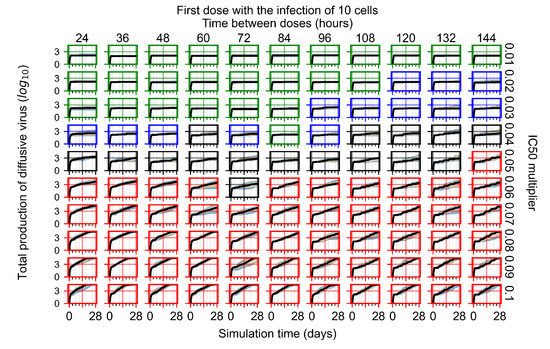
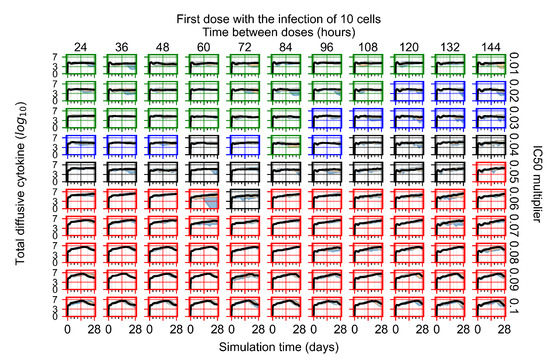
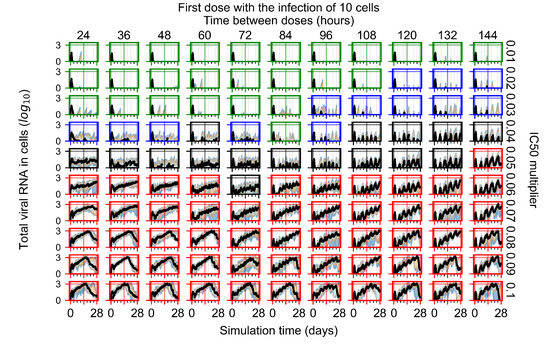
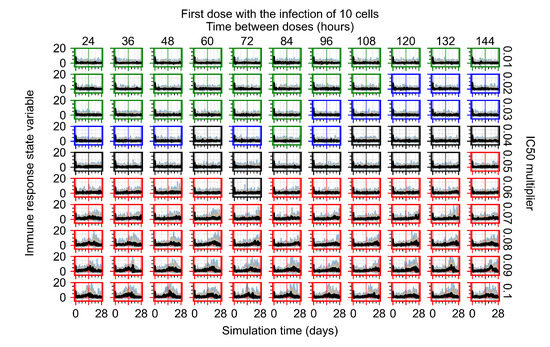
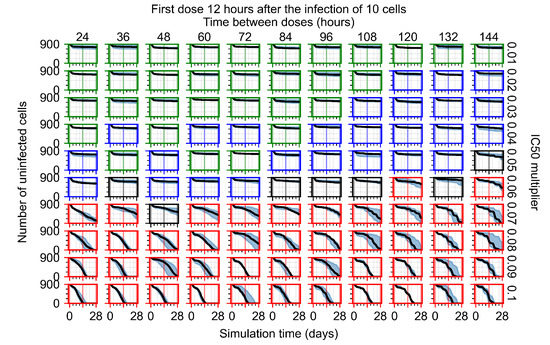
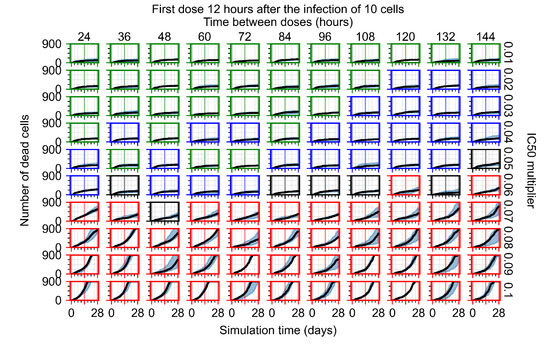
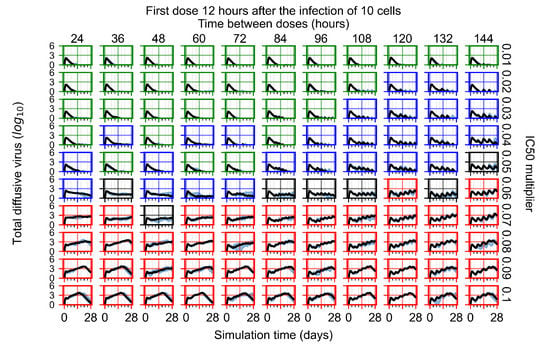
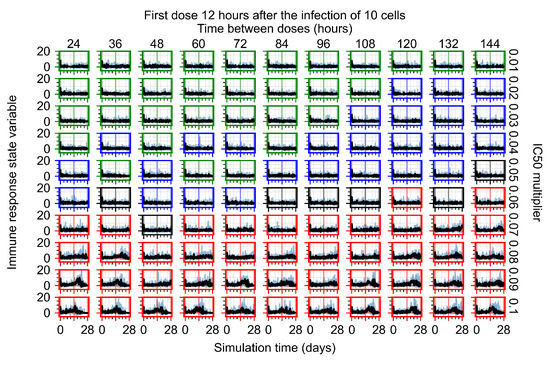
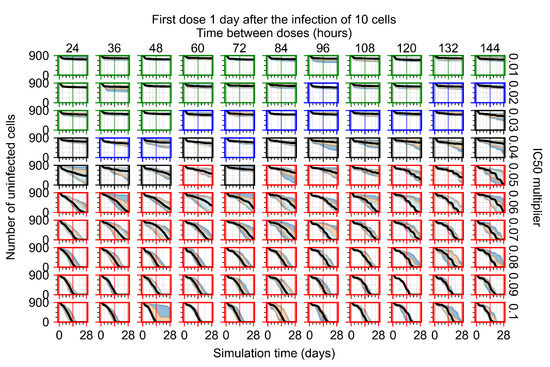
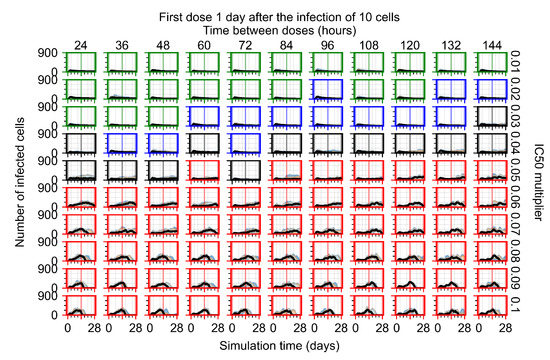
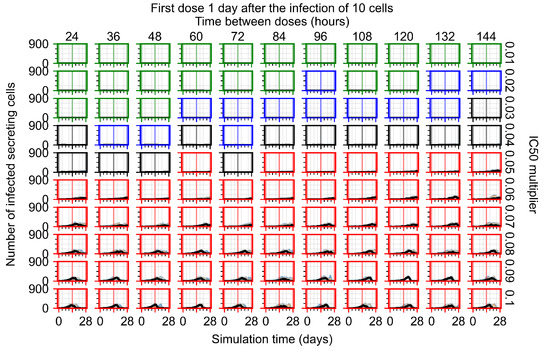
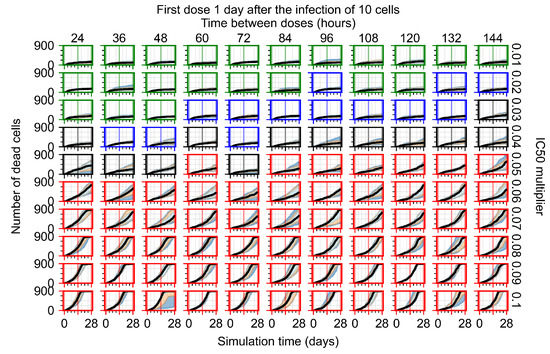
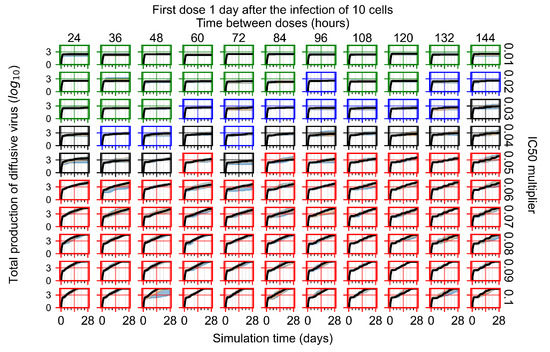
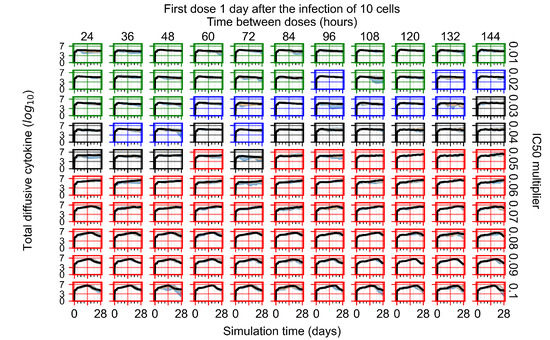
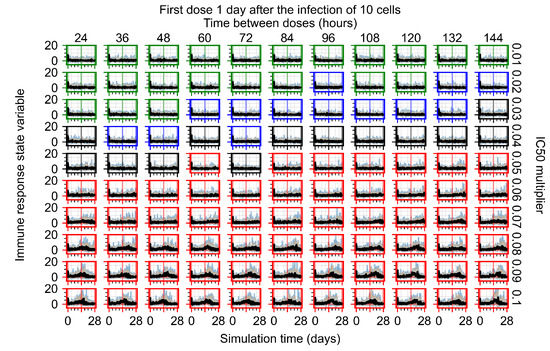
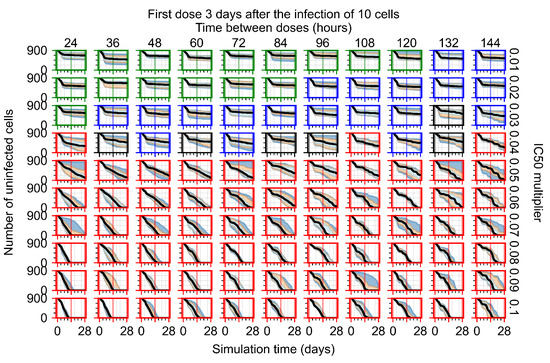
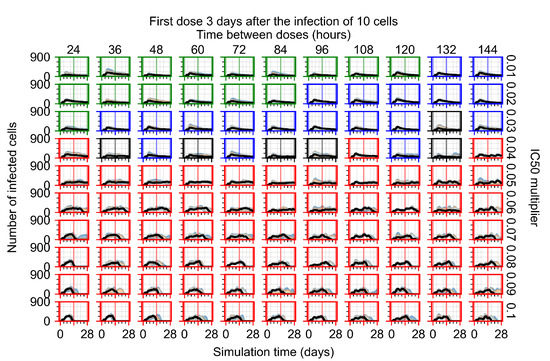
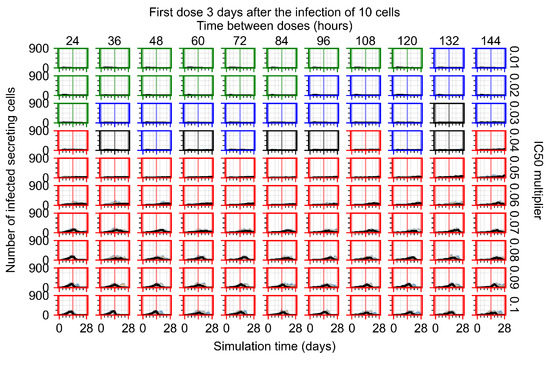
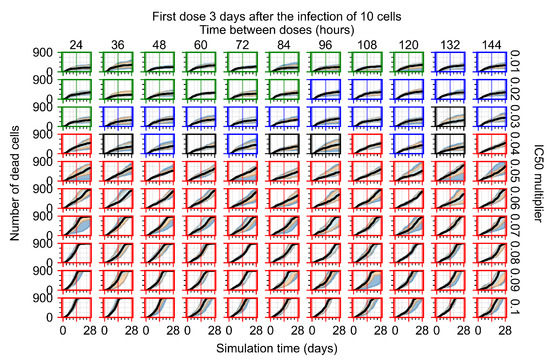
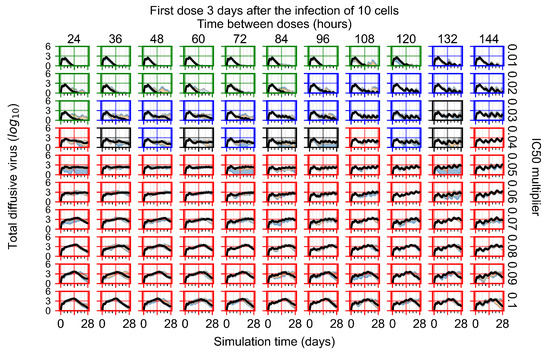
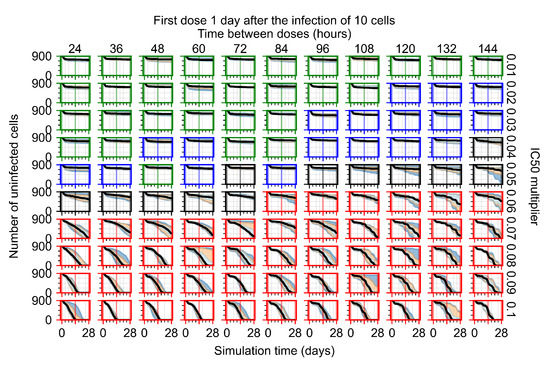
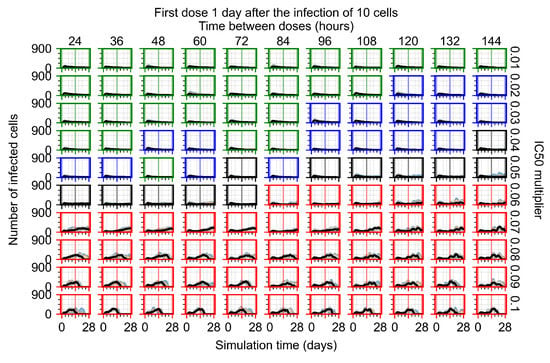
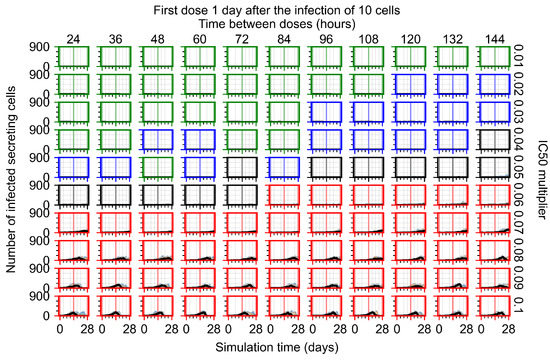
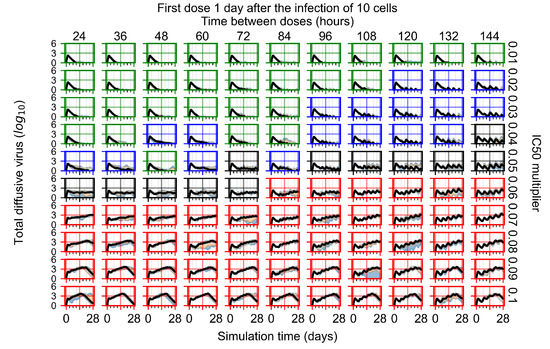
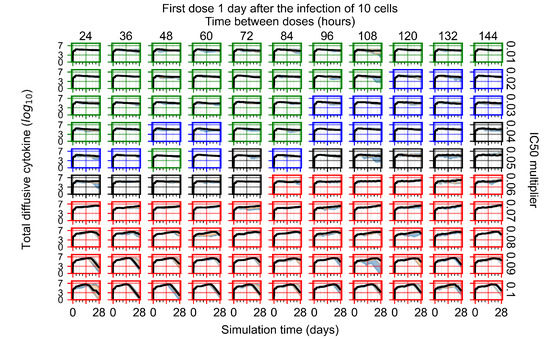
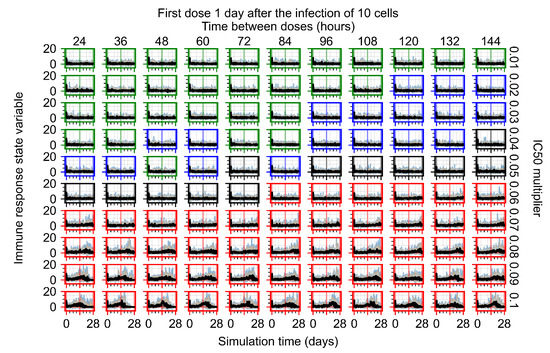
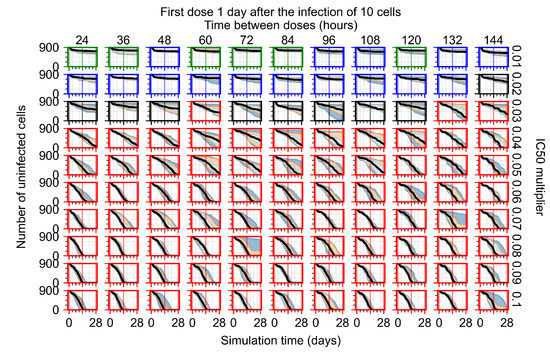
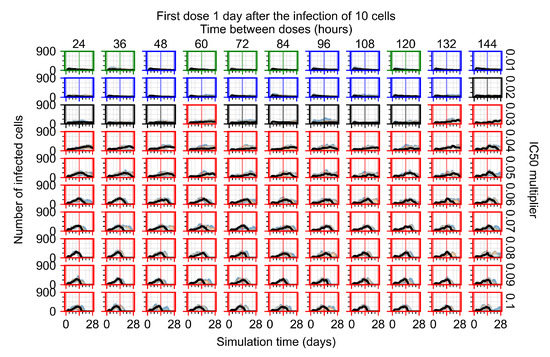
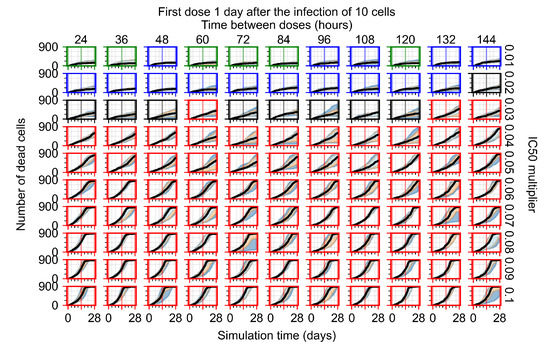
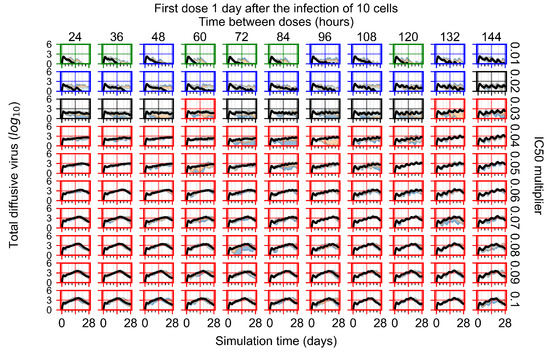
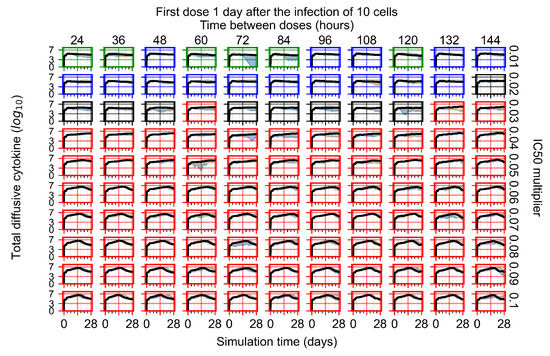
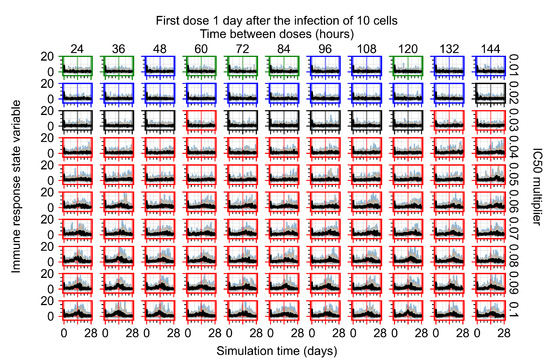
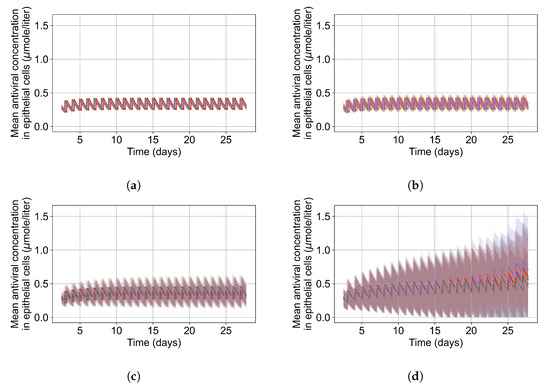
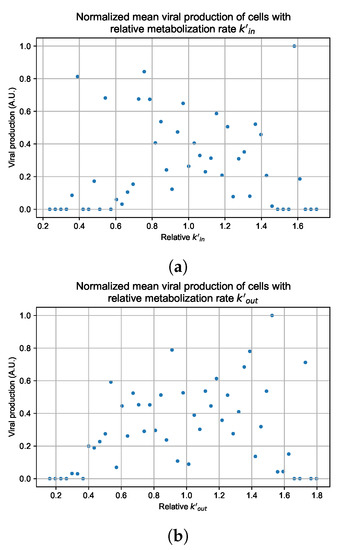
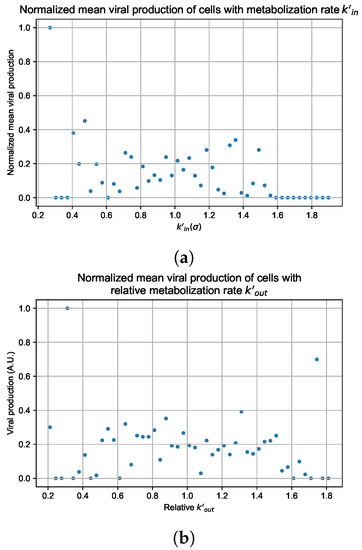
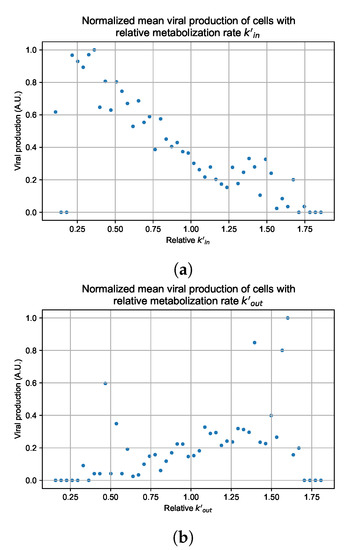
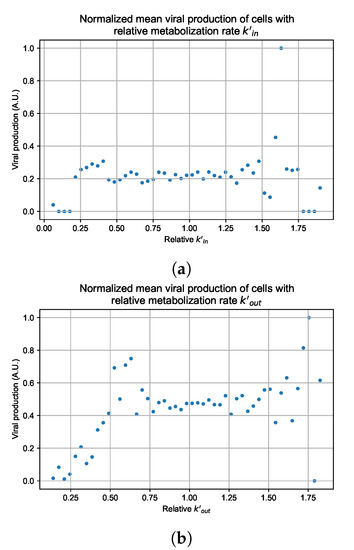
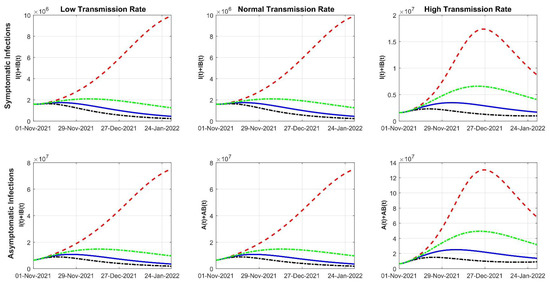
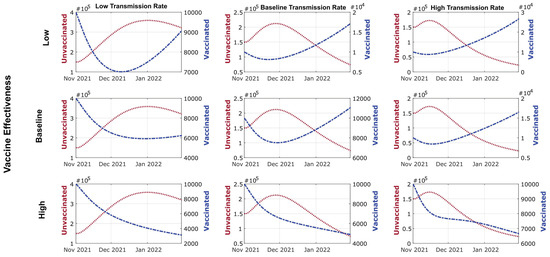
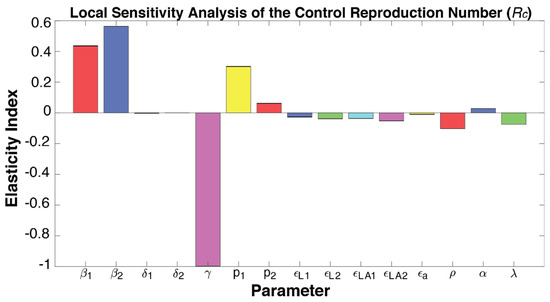
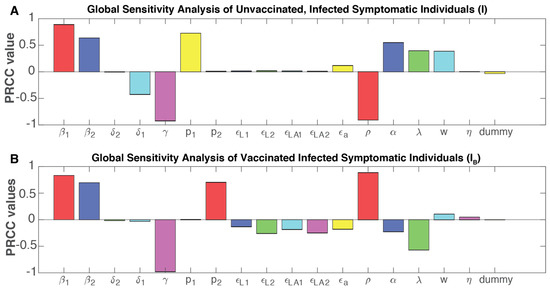
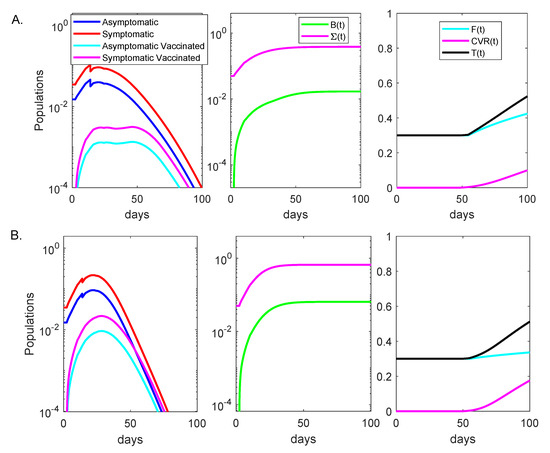
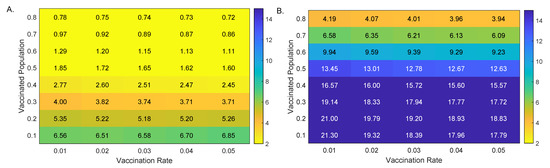
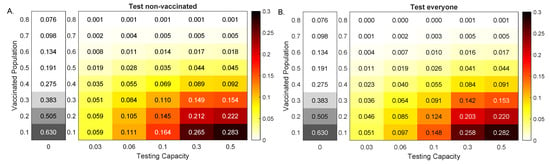
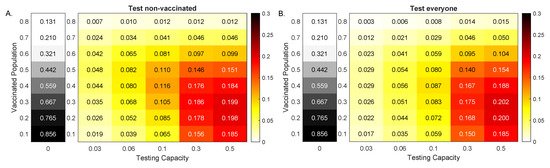
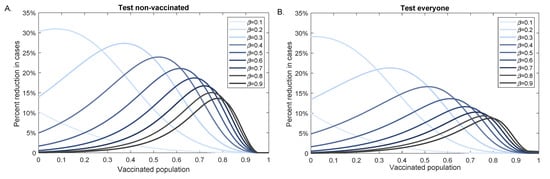
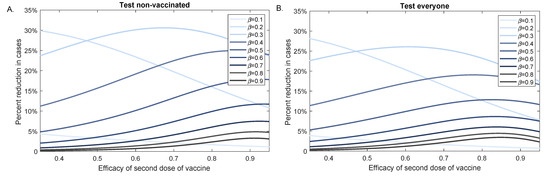
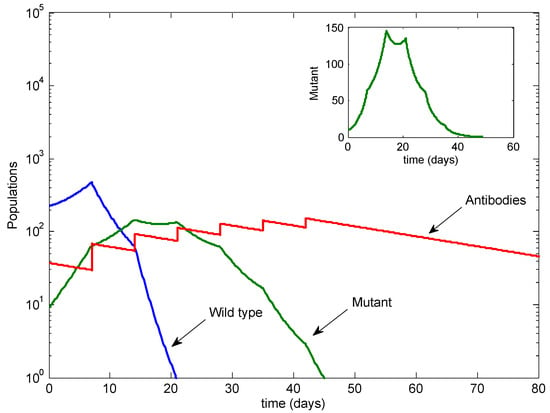
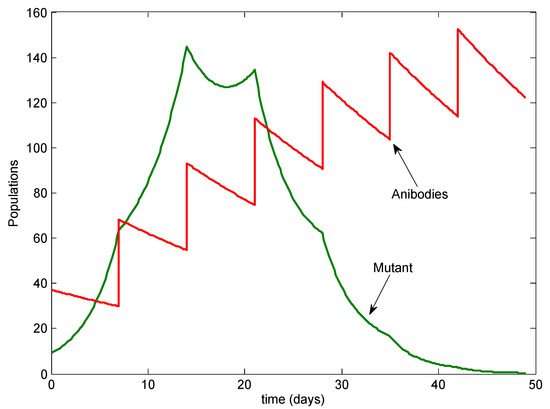
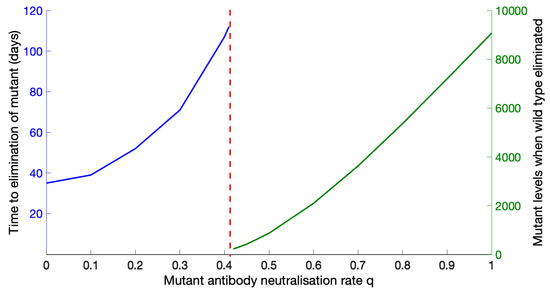
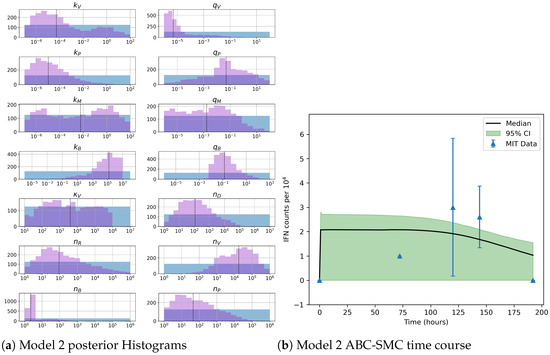
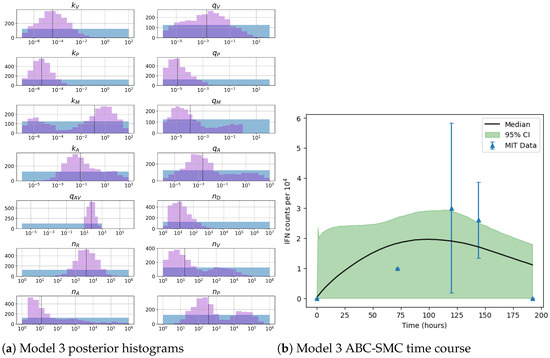
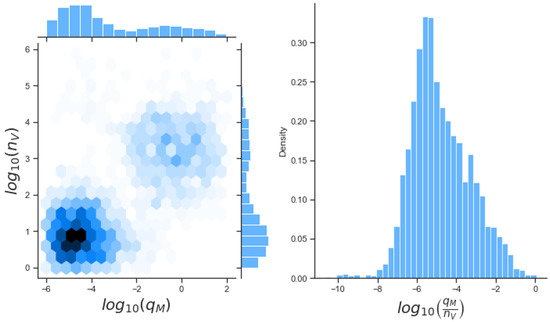
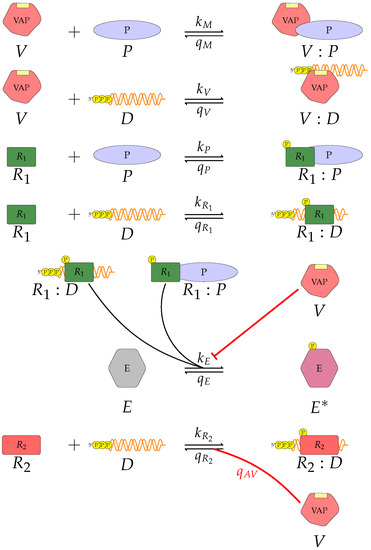
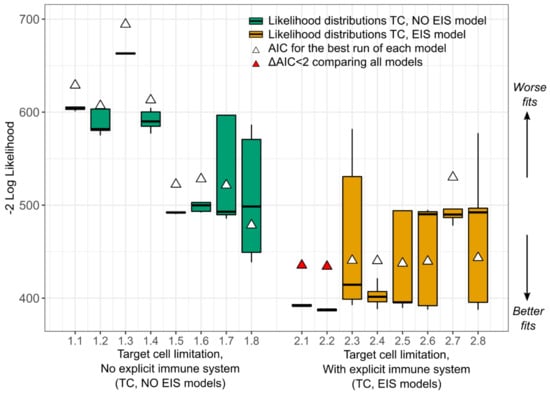
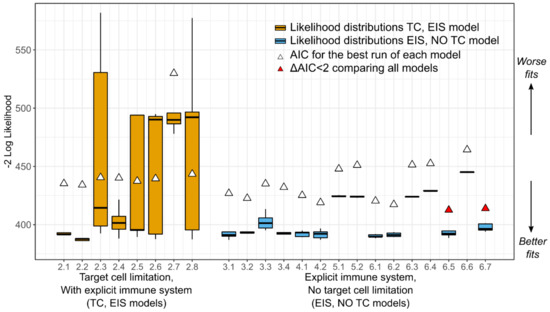
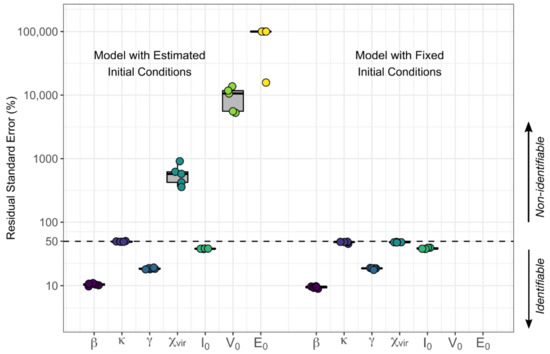
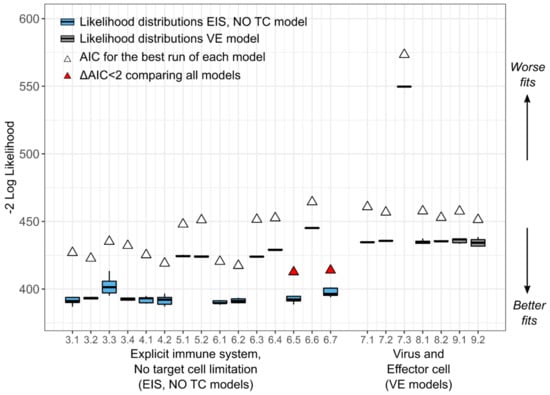
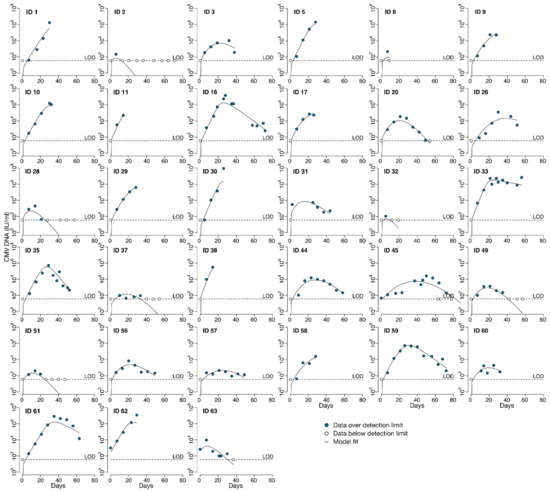
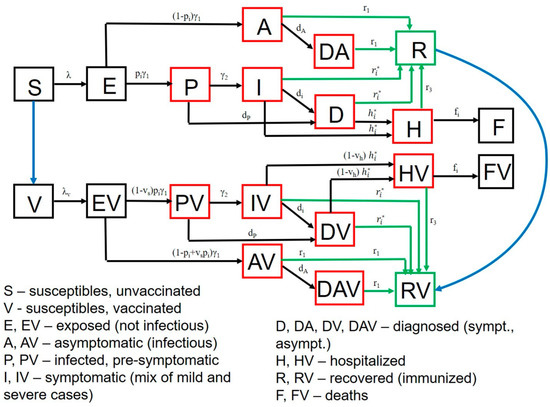
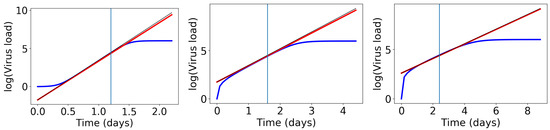
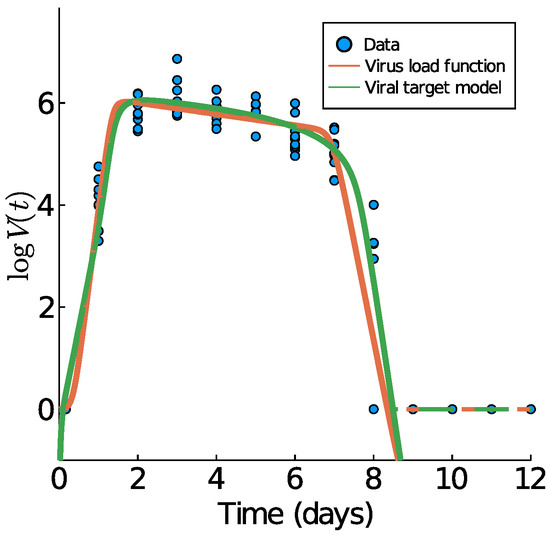
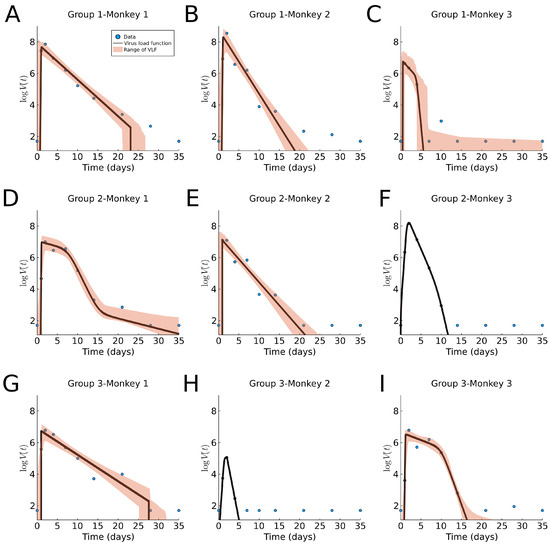
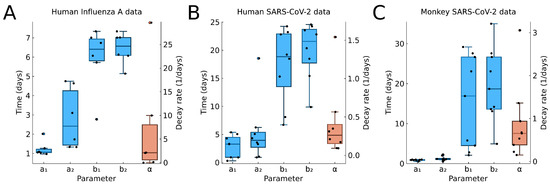
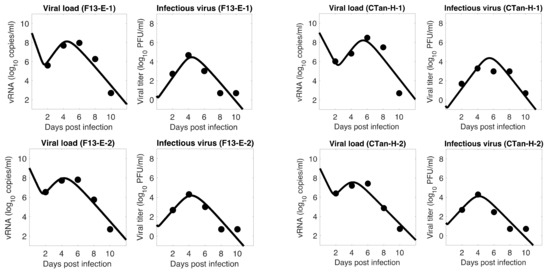
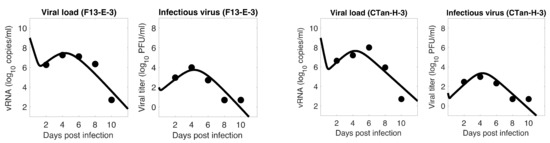
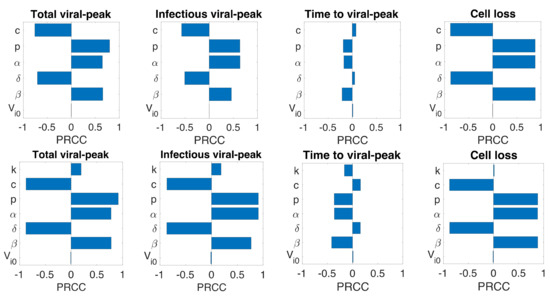
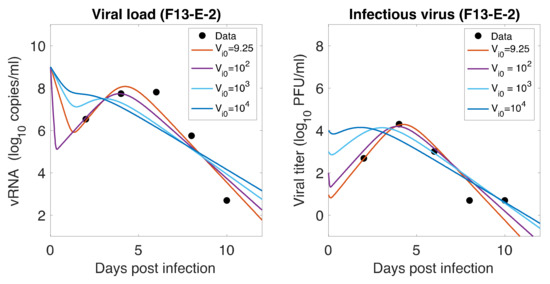
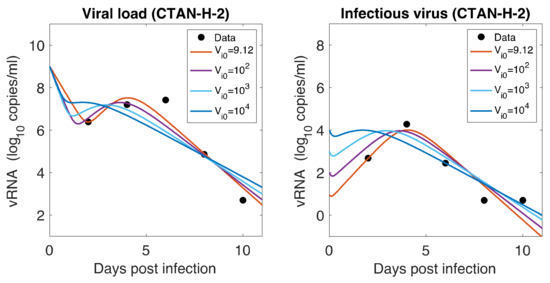
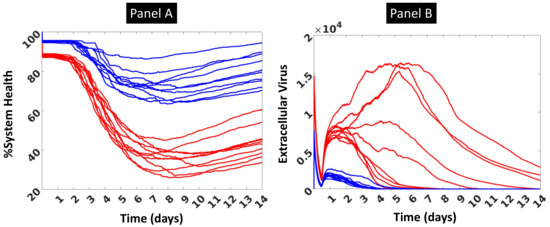
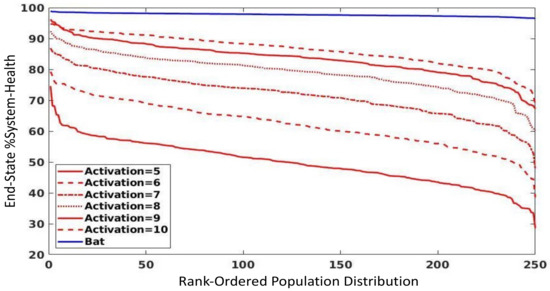
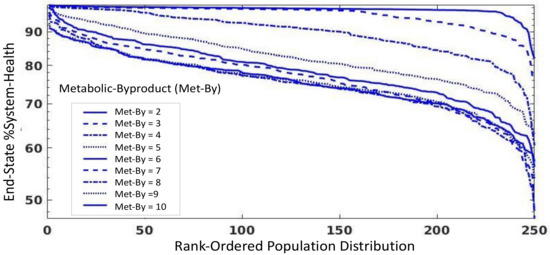
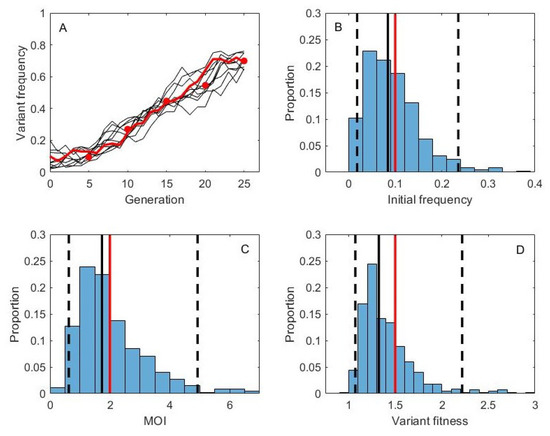
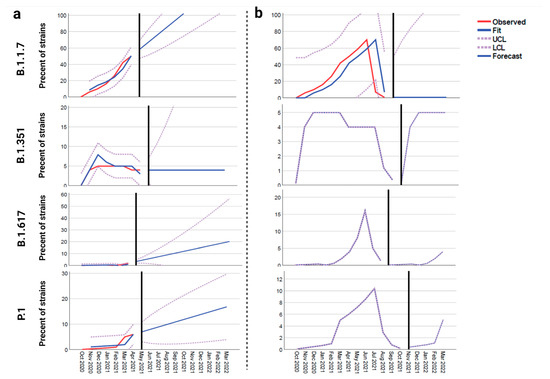
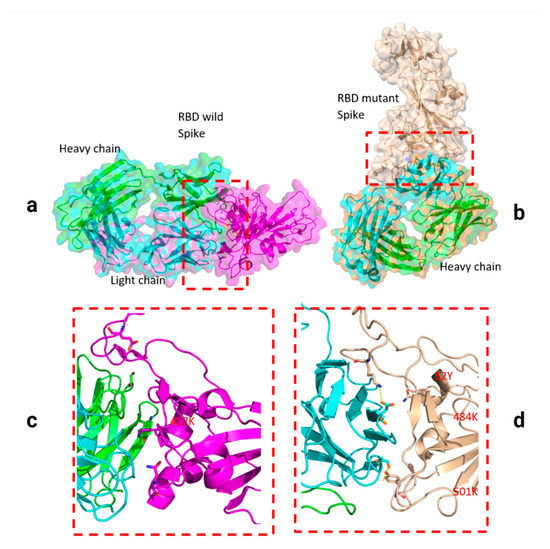
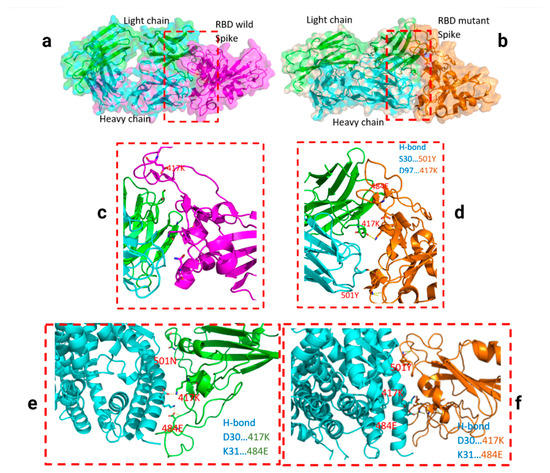
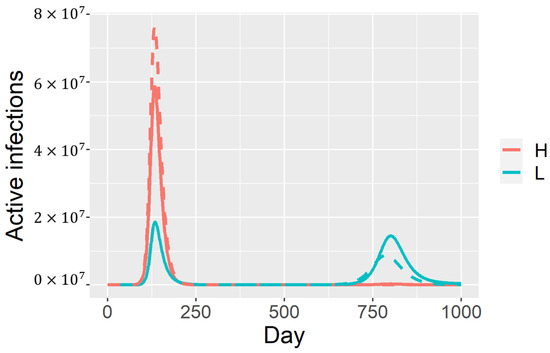
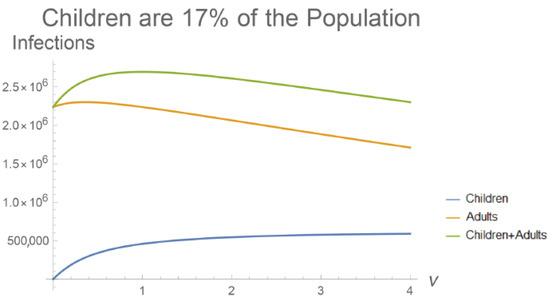
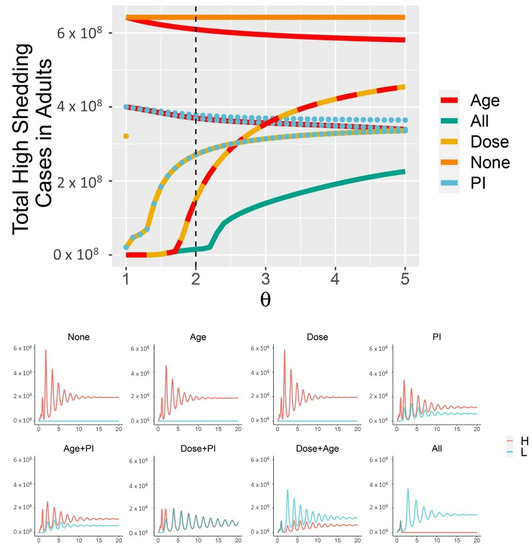
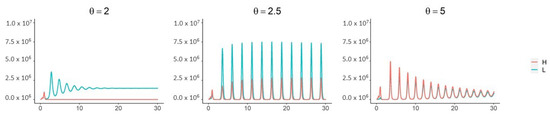
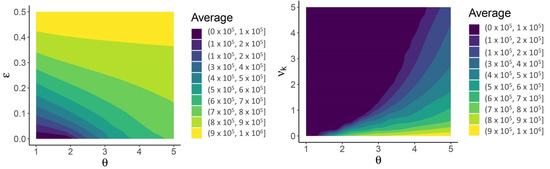
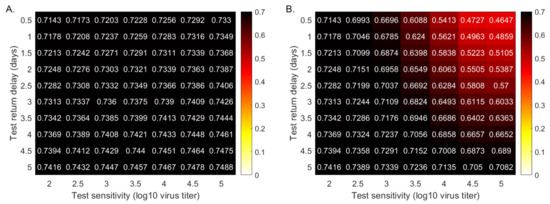
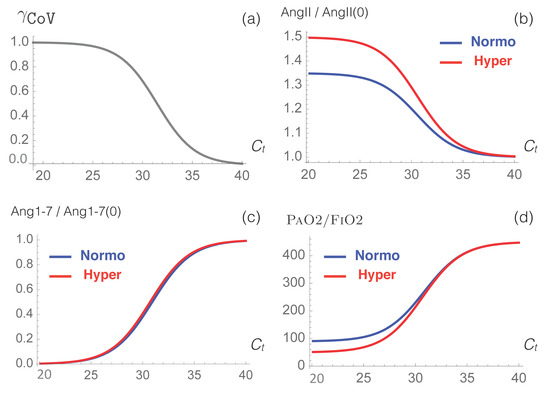
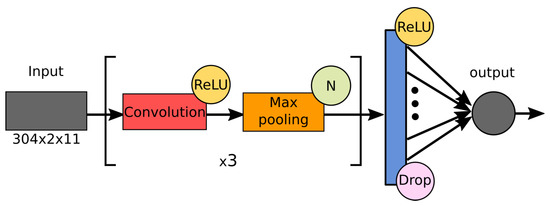
Throughout the COVID-19 pandemic, an unprecedented level of clinical nasal swab data from around the globe has been collected and shared. Positive tests have consistently revealed viral titers spanning six orders of magnitude! An open question is whether such extreme population heterogeneity is unique to SARS-CoV-2 or possibly generic to viral respiratory infections. To probe this question, we turn to the computational modeling of nasal tract infections. Employing a physiologically faithful, spatially resolved, stochastic model of respiratory tract infection, we explore the statistical distribution of human nasal infections in the immediate 48 h of infection. The spread, or heterogeneity, of the distribution derives from variations in factors within the model that are unique to the infected host, infectious variant, and timing of the test. Hypothetical factors include: (1) reported physiological differences between infected individuals (nasal mucus thickness and clearance velocity); (2) differences in the kinetics of infection, replication, and shedding of viral RNA copies arising from the unique interactions between the host and viral variant; and (3) differences in the time between initial cell infection and the clinical test. Since positive clinical tests are often pre-symptomatic and independent of prior infection or vaccination status, in the model we assume immune evasion throughout the immediate 48 h of infection. Model simulations generate the mean statistical outcomes of total shed viral load and infected cells throughout 48 h for each “virtual individual”, which we define as each fixed set of model parameters (1) and (2) above. The “virtual population” and the statistical distribution of outcomes over the population are defined by collecting clinically and experimentally guided ranges for the full set of model parameters (1) and (2). This establishes a model-generated “virtual population database” of nasal viral titers throughout the initial 48 h of infection of every individual, which we then compare with clinical swab test data. Support for model efficacy comes from the sampling of infection dynamics over the virtual population database, which reproduces the six-order-of-magnitude clinical population heterogeneity. However, the goal of this study is to answer a deeper biological and clinical question. What is the impact on the dynamics of early nasal infection due to each individual physiological feature or virus–cell kinetic mechanism? To answer this question, global data analysis methods are applied to the virtual population database that sample across the entire database and de-correlate (i.e., isolate) the dynamic infection outcome sensitivities of each model parameter. These methods predict the dominant, indeed exponential, driver of population heterogeneity in dynamic infection outcomes is the latency time of infected cells (from the moment of infection until onset of viral RNA shedding). The shedding rate of the viral RNA of infected cells in the shedding phase is a strong, but not exponential, driver of infection. Furthermore, the unknown timing of the nasal swab test relative to the onset of infection is an equally dominant contributor to extreme population heterogeneity in clinical test data since infectious viral loads grow from undetectable levels to more than six orders of magnitude within 48 h.
Full article
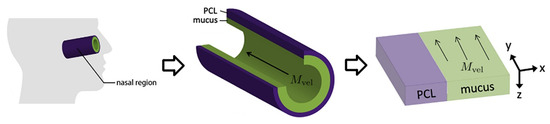
Figure 1













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}