


Viruses 2026, 18(6), 594; https://doi.org/10.3390/v18060594 (registering DOI) - 24 May 2026
Abstract
Marine algal polysaccharides have been widely investigated as antiviral candidates, yet nearly all anti-dengue studies have focused on sulfated species. Whether algal polysaccharides lacking prominent sulfation can inhibit dengue virus (DENV) remains unexplored. Here, we profiled the stage-specific antiviral activity of a heteropolysaccharide
[...] Read more.
Marine algal polysaccharides have been widely investigated as antiviral candidates, yet nearly all anti-dengue studies have focused on sulfated species. Whether algal polysaccharides lacking prominent sulfation can inhibit dengue virus (DENV) remains unexplored. Here, we profiled the stage-specific antiviral activity of a heteropolysaccharide (GLHP) from Gracilaria lemaneiformis, whose Fourier-transform infrared (FT-IR) spectrum lacks characteristic sulfate ester absorption bands, against DENV serotype 2 (DENV-2) in Huh7 and BHK-21 cells. GLHP exhibited low cytotoxicity (CC50 exceeding 1000 μg/mL in Huh7 cells and approximately 950 μg/mL in BHK-21 cells). Time-of-addition analysis revealed that co-inoculation GLHP treatment (Co-inoc.) produced the strongest and most consistent inhibition of intracellular viral RNA, whereas pre-inoculation GLHP treatment (Pre-inoc.) was ineffective, indicating that the antiviral activity is predominantly associated with the virus–cell contact and entry stage. GLHP additionally reduced extracellular progeny virus output under post-inoculation GLHP treatment (Post-inoc.) conditions, and this reduction exceeded the corresponding change in intracellular viral RNA levels, suggesting an additional effect that may involve either a late replication step or secondary entry blockade of progeny virions. Attenuation of virus-induced cytopathic effects under Co-inoc. conditions further supported the antiviral activity. To our knowledge, these findings identify GLHP as the first non-sulfated marine polysaccharide shown to exhibit stage-defined antiviral activity against DENV-2 and support further investigation of its antiviral potential and structural determinants.
Full article
(This article belongs to the Special Issue Natural, Semisynthetic, and Synthetic Antiviral Drugs: Combating Emerging and Re-Emerging Viral Threats)
►
Show Figures
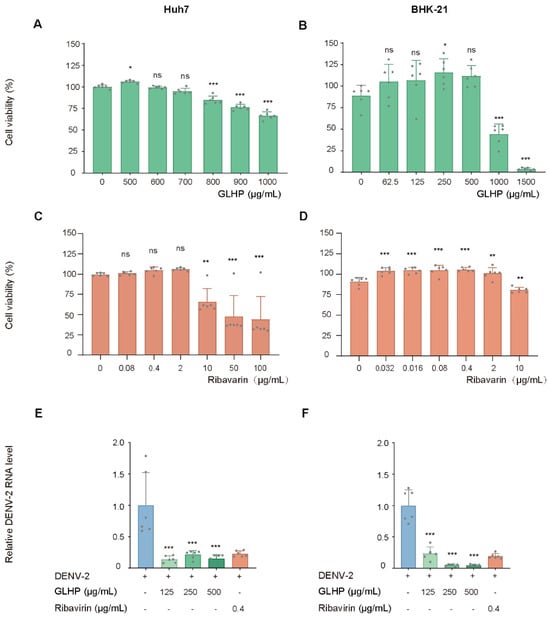
Figure 1








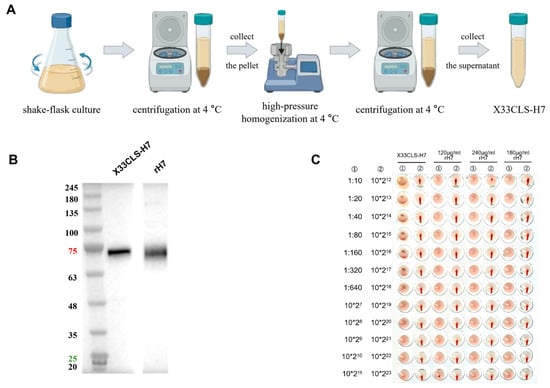










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}