- Article

The Complete Mitochondrial Genome of Eucrate alcocki (Decapoda: Brachyura: Euryplacidae) Provides New Insights Into Heterotrematan Crab Phylogeny
- Ziyang Xu,
- Jichun Li and
- Yingying Ye
- + 2 authors
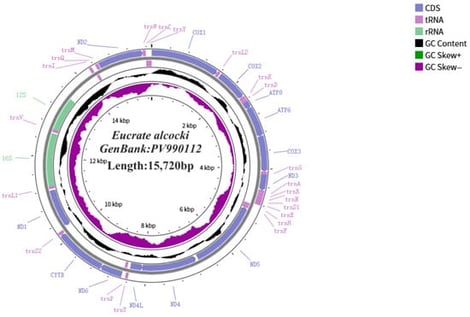
Background: This study determined the complete mitochondrial genome sequence of the marine crab to elucidate its phylogenetic position within Heterotremata, specifically the superfamily Goneplacoidea, and to explore the biological significance of its genetic composition and arrangement. Methods: The complete mitochondrial genome of Eucrate alcocki was sequenced using the Illumina platform and de novo assembled. Genome annotation and structural analysis were performed using MITOS2 and PhyloSuite. Phylogenetic relationships were reconstructed based on 13 protein-coding genes from 59 heterotrematan species using both Bayesian inference and maximum likelihood methods. Results: The mitochondrial genome of E. alcocki is a circular molecule of 15,720 bp with 72.2% AT content and a unique F-H-ND5 → H-F-ND5 gene rearrangement. Phylogenetic analysis robustly places E. alcocki in a distinct clade with Entricoplax vestita (BI = 1.00, ML = 100%), separate from the congeneric species Eucrate crenata and E. solaris, suggesting potential paraphyly within the genus Eucrate. Conclusions: This discovery provides preliminary evidence suggesting existing crab classification systems and molecular evidence for further understanding the evolutionary history of crabs. Our findings demonstrate that genomic characteristics hold significant value in revealing evolutionary pathways and can serve as a foundation for more comprehensive taxonomic and evolutionary research in the future.
7 February 2026