
ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations


Abstract
1. Introduction
2. ABCC6 and PXE
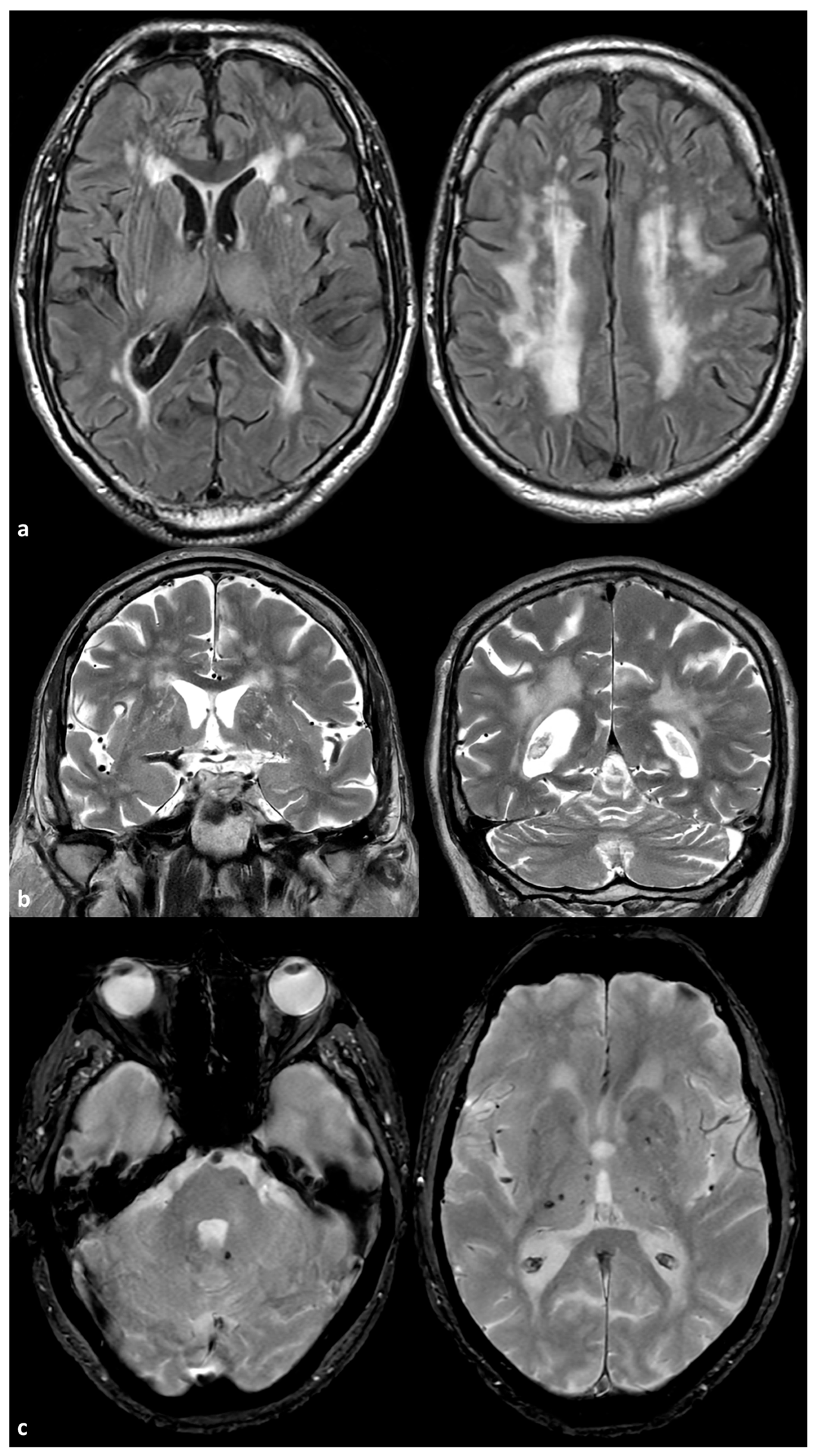
3. Small Vessel Disease Phenotype and ABCC6 Mutations
4. Mechanisms Linking ABCC6 Mutations to Small Vessel Disease
5. Potential Implications for Future Studies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Iliás, A.; Urbán, Z.; Seidl, T.L.; Le Saux, O.; Sinkó, E.; Boyd, C.D.; Sarkadi, B.; Váradi, A. Loss of ATP-dependent Transport Activity in Pseudoxanthoma Elasticum-associated Mutants of Human ABCC6 (MRP6). J. Biol. Chem. 2002, 277, 16860–16867. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, O.; Martin, L.; Aherrahrou, Z.; Leftheriotis, G.; Váradi, A.; Brampton, C.N. The molecular and physiological roles of ABCC6: More than meets the eye. Front. Genet. 2012, 3, 289. [Google Scholar] [CrossRef]
- Le Saux, O.; Urban, Z.; Tschuch, C.; Csiszar, K.; Bacchelli, B.; Quaglino, D.; Pasquali-Ronchetti, I.; Pope, F.M.; Richards, A.; Terry, S.; et al. Mutations in a gene encoding an ABC transporter cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 223–227. [Google Scholar] [CrossRef]
- Jansen, R.S.; Küçükosmanoğlu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.M.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Yegutkin, G.G.; Khiati, S.; Pomozi, V.; Le Saux, O.; Leftheriotis, G.; Lenaers, G.; Henrion, D.; Martin, L. Alteration of Extracellular Nucleotide Metabolism in Pseudoxanthoma Elasticum. J. Investig. Dermatol. 2018, 138, 1862–1870. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, S.G.; Ferreira, C.R.; MacFarlane, E.G.; Riddle, R.C.; Tomlinson, R.E.; Chew, E.Y.; Martin, L.; Ma, C.-T.; Sergienko, E.; Pinkerton, A.B.; et al. Ectopic calcification in pseudoxanthoma elasticum responds to inhibition of tissue-nonspecific alkaline phosphatase. Sci. Transl. Med. 2017, 9, eaal1669. [Google Scholar] [CrossRef] [PubMed]
- Orriss, I.R.; Arnett, T.R.; Russell, R.G.G. Pyrophosphate: A key inhibitor of mineralisation. Curr. Opin. Pharmacol. 2016, 28, 57–68. [Google Scholar] [CrossRef]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Elferink, R.P.O.; Borst, P.; et al. ABCC6–Mediated ATP Secretion by the Liver Is the Main Source of the Mineralization Inhibitor Inorganic Pyrophosphate in the Systemic Circulation—Brief Report. Arter. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Pomozi, V.; Brampton, C.; van de Wetering, K.; Zoll, J.; Calio, B.; Pham, K.; Owens, J.B.; Marh, J.; Moisyadi, S.; Váradi, A.; et al. Pyrophosphate Supplementation Prevents Chronic and Acute Calcification in ABCC6-Deficient Mice. Am. J. Pathol. 2017, 187, 1258–1272. [Google Scholar] [CrossRef]
- Beck, K.; Hayashi, K.; Dang, K.; Hayashi, M.; Boyd, C.D. Analysis of ABCC6 (MRP6) in normal human tissues. Histochem. Cell. Biol. 2005, 123, 517–528. [Google Scholar] [CrossRef]
- Beck, K.; Hayashi, K.; Nishiguchi, B.; Le Saux, O.; Hayashi, M.; Boyd, C.D. The Distribution of Abcc6 in Normal Mouse Tissues Suggests Multiple Functions for this ABC Transporter. J. Histochem. Cytochem. 2003, 51, 887–902. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.-F.; et al. Generalized Arterial Calcification of Infancy and Pseudoxanthoma Elasticum Can Be Caused by Mutations in Either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef]
- Hamlin, N.; Beck, K.; Bacchelli, B.; Cianciulli, P.; Pasquali-Ronchetti, I.; Le Saux, O. Acquired Pseudoxanthoma elasticum-like syndrome in beta-thalassaemia patients. Br. J. Haematol. 2003, 122, 852–854. [Google Scholar] [CrossRef]
- Martin, L.; Douet, V.; VanWart, C.M.; Heller, M.B.; Le Saux, O. A Mouse Model of β-Thalassemia Shows a Liver-Specific Down-Regulation of Abcc6 Expression. Am. J. Pathol. 2011, 178, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Rios-Montenegro, E.N.; Behrens, M.M.; Hoyt, W.F. Pseudoxanthoma elasticum. Arch. Neurol. 1972, 26, 151–155. [Google Scholar] [CrossRef]
- Yasuhara, T.; Sugiu, K.; Kakishita, M.; Date, I. Pseudoxanthoma elasticum with carotid rete mirabile. Clin. Neurol. Neurosurg. 2004, 106, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Del Zotto, E.; Ritelli, M.; Pezzini, A.; Drera, B.; Gamba, M.; Giossi, A.; Volonghi, I.; Costa, P.; Barlati, S.; Gasparotti, R.; et al. Clinical, neuroradiological and molecular features of a patient affected by pseudoxhantoma elasticum associated to carotid rete mirabile: Case report. Clin. Neurol. Neurosurg. 2012, 114, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Prick, J.; Thijssen, H. Radiodiagnostic signs in pseudoxanthoma elasticum generalisatum (dysgenesis elastofibrillaris mineralisans). Clin. Radiol. 1977, 28, 549–554. [Google Scholar] [CrossRef]
- Iqbal, A.; Alter, M.; Lee, S.H. Pseudoxanthoma elasticum: A review of neurological complications. Ann. Neurol. 1978, 4, 18–20. [Google Scholar] [CrossRef]
- Dixon, J.M. Angioid streaks and pseudoxanthoma elasticum with aneurysm of the internal carotid artery. Am. J. Ophthalmol. 1951, 34, 1322–1323. [Google Scholar] [CrossRef]
- Scheie, H.G.; Hogan, T.F. Angioid Streaks and Generalized Arterial Disease. Arch. Ophthalmol. 1957, 57, 855–868. [Google Scholar] [CrossRef]
- Munyer, T.; Margulis, A. Pseudoxanthoma elasticum with internal carotid artery aneurysm. Am. J. Roentgenol. 1981, 136, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Kito, K.; Kobayashi, N.; Mori, N.; Kohno, H. Ruptured aneurysm of the anterior spinal artery associated with pseudoxanthoma elasticum. J. Neurosurg. 1983, 58, 126–128. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.G.K.; Beohar, P.C.; Ghosh, S.K.; Gupta, P.S. Subarachnoid haemorrhage in pseudoxanthoma elasticum. Postgrad. Med. J. 1974, 50, 774–776. [Google Scholar] [CrossRef]
- Bock, A.; Schwegler, G. Intracerebral haemorrhage as first manifestation of Pseudoxanthoma elasticum. Clin. Neurol. Neurosurg. 2008, 110, 262–264. [Google Scholar] [CrossRef]
- Pavlovic, A.M.; Zidverc-Trajkovic, J.; Milovic, M.M.; Pavlovic, D.M.; Jovanovic, Z.; Mijajlovic, M.; Petrovic, M.; Kostic, V.S.; Sternic, N. Cerebral Small Vessel Disease in Pseudoxanthoma Elasticum: Three Cases. Can. J. Neurol. Sci./J. Can. Sci. Neurol. 2005, 32, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Dalloz, M.-A.; Debs, R.; Bensa, C.; Alamowitch, S. Hypersignaux de la substance blanche révélant un pseudoxanthome élastique. Rev. Neurol. 2010, 166, 844–848. [Google Scholar] [CrossRef]
- Hirano, T.; Hashimoto, Y.; Kimura, K.; Uchino, M. Lacunar brain infarction in patients with pseudoxanthoma elasticum. Rinsho Shinkeigaku 1996, 36, 633–639. [Google Scholar]
- Germain, D.P.; Perdu, J. Identification of novel polymorphisms in the pM5 and MRP1 (ABCC1) genes at locus 16p13.1 and exclusion of both genes as responsible for pseudoxanthoma elasticum. Hum. Mutat. 2001, 17, 74–75. [Google Scholar] [CrossRef]
- Bergen, A.A.; Plomp, A.S.; Schuurman, E.J.; Terry, S.; Breuning, M.; Dauwerse, H.; Swart, J.; Kool, M.; van Soest, S.; Baas, F.; et al. Mutations in ABCC6 cause pseudoxanthoma elasticum. Nat. Genet. 2000, 25, 228–231. [Google Scholar] [CrossRef]
- Ringpfeil, F.; Lebwohl, M.G.; Christiano, A.M.; Uitto, J. Pseudoxanthoma elasticum: Mutations in the MRP6 gene encoding a transmembrane ATP-binding cassette (ABC) transporter. Proc. Natl. Acad. Sci. USA 2000, 97, 6001–6006. [Google Scholar] [CrossRef]
- Germain, D.P.; Perdu, J.; Remones, V.; Jeunemaitre, X. Homozygosity for the R1268Q Mutation in MRP6, the Pseudoxanthoma Elasticum Gene, Is Not Disease-Causing. Biochem. Biophys. Res. Commun. 2000, 274, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Struk, B.; Cai, L.; Zäch, S.; Ji, W.; Chung, J.; Lumsden, A.; Stumm, M.; Huber, M.; Schaen, L.; Kim, C.-A.; et al. Mutations of the gene encoding the transmembrane transporter protein ABC-C6 cause pseudoxanthoma elasticum. J. Mol. Med. 2000, 78, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tiwari, A.K. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011, 278, 3226–3245. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, K.; Barna, L.; Symmons, O.; Závodszky, P.; Váradi, A. Clustering of disease-causing mutations on the domain–domain interfaces of ABCC6. Biochem. Biophys. Res. Commun. 2009, 379, 706–709. [Google Scholar] [CrossRef]
- Arányi, T.; Bacquet, C.; de Boussac, H.; Ratajewski, M.; Pomozi, V.; Fülöp, K.; Brampton, C.N.; Pulaski, L.; Le Saux, O.; Váradi, A.; et al. Transcriptional regulation of the ABCC6 gene and the background of impaired function of missense disease-causing mutations. Front. Genet. 2013, 4, 39319. [Google Scholar] [CrossRef]
- Ratajewski, M.; de Boussac, H.; Sachrajda, I.; Bacquet, C.; Kovács, T.; Váradi, A.; Pulaski, L.; Arányi, T. ABCC6 Expression Is Regulated by CCAAT/Enhancer-Binding Protein Activating a Primate-Specific Sequence Located in the First Intron of the Gene. J. Investig. Dermatol. 2012, 132, 2709–2717. [Google Scholar] [CrossRef] [PubMed]
- de Boussac, H.; Ratajewski, M.; Sachrajda, I.; Köblös, G.; Tordai, A.; Pulaski, L.; Buday, L.; Váradi, A.; Arányi, T. The ERK1/2-hepatocyte nuclear factor 4alpha axis regulates human ABCC6 gene expression in hepatocytes. J. Biol. Chem. 2010, 285, 22800–22808. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, M.G.; Chen, Z.-S.; Shchaveleva, I.; Zeng, H.; Kruh, G.D. Characterization of the drug resistance and transport properties of multidrug resistance protein 6 (MRP6, ABCC6). Cancer Res. 2002, 62, 6172–6177. [Google Scholar]
- Miksch, S.; Lumsden, A.; Guenther, U.P.; Foernzler, D.; Christen-Zäch, S.; Daugherty, C.; Ramesar, R.K.S.; Lebwohl, M.; Hohl, D.; Neldner, K.H.; et al. Molecular genetics of pseudoxanthoma elasticum: Type and frequency of mutations in ABCC6. Hum. Mutat. 2005, 26, 235–248. [Google Scholar] [CrossRef]
- Christen-Zach, S.; Huber, M.; Struk, B.; Lindpaintner, K.; Munier, F.; Panizzon, R.G.; Hohl, D. Pseudoxanthoma elasticum: Evaluation of diagnostic criteria based on molecular data. Br. J. Dermatol. 2006, 155, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Pfendner, E.G.; Vanakker, O.M.; Terry, S.F.; Vourthis, S.; McAndrew, P.E.; McClain, M.R.; Fratta, S.; Marais, A.-S.; Hariri, S.; Coucke, P.J.; et al. Mutation detection in the ABCC6 gene and genotype phenotype analysis in a large international case series affected by pseudoxanthoma elasticum. J. Med. Genet. 2007, 44, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Hornstrup, L.S.; Tybjærg-Hansen, A.; Haase, C.L.; Nordestgaard, B.G.; Sillesen, H.; Grande, P.; Frikke-Schmidt, R. Heterozygosity for R1141X in ABCC6 and Risk of Ischemic Vascular Disease. Circ. Cardiovasc. Genet. 2011, 4, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Akoglu, G.; Li, Q.; Gokoz, O.; Gazyagci, A.S.; Uitto, J. Clinical and histopathological characteristics of a family with R1141X mutation of pseudoxanthoma elasticum—Presymptomatic testing and lack of carrier phenotypes. Int. J. Dermatol. 2014, 53, 692–698. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Leroy, B.P.; Coucke, P.; Bercovitch, L.G.; Uitto, J.; Viljoen, D.; Terry, S.F.; Van Acker, P.; Matthys, D.; Loeys, B.; et al. Novel clinico-molecular insights in pseudoxanthoma elasticum provide an efficient molecular screening method and a comprehensive diagnostic flowchart. Hum. Mutat. 2008, 29, 205. [Google Scholar] [CrossRef]
- Chassaing, N.; Martin, L.; Calvas, P.; Le Bert, M.; Hovnanian, A. Pseudoxanthoma elasticum: A clinical, pathophysiological and genetic update including 11 novel ABCC6 mutations. J. Med. Genet. 2005, 42, 881–892. [Google Scholar] [CrossRef]
- Li, Q.; Arányi, T.; Váradi, A.; Terry, S.F.; Uitto, J. Research Progress in Pseudoxanthoma Elasticum and Related Ectopic Mineralization Disorders. J. Investig. Dermatol. 2016, 136, 550–556. [Google Scholar] [CrossRef]
- Kuzaj, P.; Kuhn, J.; Michalek, R.D.; Karoly, E.D.; Faust, I.; Dabisch-Ruthe, M.; Knabbe, C.; Hendig, D.; Dardis, A. Large-Scaled Metabolic Profiling of Human Dermal Fibroblasts Derived from Pseudoxanthoma Elasticum Patients and Healthy Controls. PLoS ONE 2014, 9, e108336. [Google Scholar] [CrossRef]
- Jiang, Q.; Oldenburg, R.; Otsuru, S.; Grand-Pierre, A.E.; Horwitz, E.M.; Uitto, J. Parabiotic heterogenetic pairing of Abcc6−/−/Rag1−/− mice and their wild-type counterparts halts ectopic mineralization in a murine model of pseudoxanthoma elasticum. Am. J. Pathol. 2010, 176, 1855–1862. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Blum, R.R.; Singer, G.K.; Stern, D.K.; Emanuel, P.O.; Fuchs, W.; Phelps, R.G.; Terry, S.F.; Lebwohl, M.G. A randomized controlled trial of oral phosphate binders in the treatment of pseudoxanthoma elasticum. J. Am. Acad. Dermatol. 2011, 65, 341–348. [Google Scholar] [CrossRef]
- Hendig, D.; Schulz, V.; Arndt, M.; Szliska, C.; Kleesiek, K.; Gotting, C. Role of serum fetuin-A, a major inhibitor of systemic calcifi-cation, in pseudoxanthoma elasticum. Clin. Chem. 2006, 52, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Suliman, M.E.; Garcia-Lopez, E.; Anderstam, B.; Lindholm, B.; Stenvinkel, P. Vascular calcification inhibitors in relation to cardiovascular disease with special emphasis on fetuin-A in chronic kidney disease. Adv. Clin. Chem. 2008, 46, 217–262. [Google Scholar] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Lau, W.L.; Liu, S.; Vaziri, N.D. Chronic Kidney Disease Results in Deficiency of ABCC6, the Novel Inhibitor of Vascular Calcification. Am. J. Nephrol. 2014, 40, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development 2015, 142, 1095–1101. [Google Scholar] [CrossRef]
- Kariminejad, A.; Bozorgmehr, B.; Najafi, A.; Khoshaeen, A.; Ghalandari, M.; Najmabadi, H.; Kariminejad, M.H.; Vanakker, O.M.; Hosen, M.J.; Malfait, F.; et al. Retinitis pigmentosa, cutis laxa, and pseudoxanthoma elasticum-like skin manifestations associated with GGCX mutations. J. Investig. Dermatol. 2014, 134, 2331–2338. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, Q.; Grand-Pierre, A.E.; Schurgers, L.J.; Uitto, J. Administration of vitamin K does not counteract the ectopic minerali-zation of connective tissues in Abcc6 (−/−) mice, a model for pseudoxanthoma elasticum. Cell Cycle 2011, 10, 701–707. [Google Scholar] [CrossRef]
- Brampton, C.; Yamaguchi, Y.; Vanakker, O.; Van Laer, L.; Chen, L.-H.; Thakore, M.; De Paepe, A.; Pomozi, V.; Szabó, P.T.; Martin, L.; et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 2011, 10, 1810–1820. [Google Scholar] [CrossRef]
- Gorgels, T.G.M.F.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125–1135. [Google Scholar] [CrossRef]
- St Hilaire, C.; Ziegler, S.G.; Markello, T.C.; Brusco, A.; Groden, C.; Gill, F.; Carlson-Donohoe, H.; Lederman, R.J.; Chen, M.Y.; Yang, D.; et al. NT5E mutations and arterial calcifications. N. Engl. J. Med. 2011, 364, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Markello, T.C.; Pak, L.K.; Hilaire, C.S.; Dorward, H.; Ziegler, S.G.; Chen, M.Y.; Chaganti, K.; Nussbaum, R.L.; Boehm, M.; Gahl, W.A. Vascular pathology of medial arterial calcifications in NT5E deficiency: Implications for the role of adenosine in pseudoxanthoma elasticum. Mol. Genet. Metab. 2011, 103, 44–50. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.; Vanakker, O.M. From variome to phenome: Pathogenesis, diagnosis and management of ectopic mineralization disorders. World J. Clin. Cases 2015, 3, 556–574. [Google Scholar] [CrossRef]
- Li, Q.; Price, T.P.; Sundberg, J.P.; Uitto, J. Juxta-articular joint-capsule mineralization in CD73 deficient mice: Similarities to patients with NT5E mutations. Cell Cycle 2014, 13, 2609–2615. [Google Scholar] [CrossRef] [PubMed]
- Szabó, Z.; Váradi, A.; Li, Q.; Uitto, J. ABCC6 does not transport adenosine—Relevance to pathomechanism of pseudoxanthoma elasticum. Mol. Genet. Metab. 2011, 104, 421. [Google Scholar] [CrossRef]
- Garcia-Fernandez, M.I.; Gheduzzi, D.; Boraldi, F.; Paolinelli, C.D.; Sanchez, P.; Valdivielso, P.; Morilla, M.J.; Quaglino, D.; Guerra, D.; Casolari, S.; et al. Parameters of oxidative stress are present in the circulation of PXE patients. Biochim. et Biophys. Acta (BBA)—Mol. Basis Dis. 2008, 1782, 474–481. [Google Scholar] [CrossRef]
- Voskou, S.; Aslan, M.; Fanis, P.; Phylactides, M.; Kleanthous, M. Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol. 2015, 6, 226–239. [Google Scholar] [CrossRef]
- Baccarani-Contri, M.; Bacchelli, B.; Boraldi, F.; Quaglino, D.; Taparelli, F.; Carnevalia, E.; Francomano, M.A.; Seidenari, S.; Bettoli, V.; De Sanctis, V.; et al. Characterization of pseudoxanthoma elasticum-like lesions in the skin of patients with ?-thalassemia. J. Am. Acad. Dermatol. 2001, 44, 33–39. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Pfendner, E.; Váradi, A.; Uitto, J. Pseudoxanthoma elasticum: Clinical phenotypes, molecular genetics and putative pathomechanisms. Exp Dermatol. 2009, 18, 1–11. [Google Scholar] [CrossRef]
- Fabbri, E.; Forni, G.L.; Guerrini, G.; Borgna-Pignatti, C. Pseudoxanthoma elasticum-like syndrome and thalassemia: An update. Dermatol. Online J. 2009, 15, 7. [Google Scholar] [CrossRef]
- Martin, L.J.; Lau, E.; Singh, H.; Vergnes, L.; Tarling, E.J.; Mehrabian, M.; Mungrue, I.; Xiao, S.; Shih, D.; Castellani, L.; et al. ABCC6 localizes to the mitochondria-associated mem-brane. Circ. Res. 2012, 111, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Ferré, M.; Reynier, P.; Chevrollier, A.; Prunier-Mirebeau, D.; Lefthériotis, G.; Henrion, D.; Bonneau, D.; Procaccio, V.; Martin, L. Is ABCC6 a genuine mitochondrial protein? BMC Res Notes. 2013, 6, 427. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.S.M.; Moestrup, S.K. Is classical pseudoxanthoma elasticum a consequence of hepatic ‘intoxication’ due to ABCC6 substrate accumulation in the liver? Expert Rev. Endocr. Metabol. 2013, 8, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Frank, M.; Thisse, C.I.; Thisse, B.V.; Uitto, J. Zebrafish: A Model System to Study Heritable Skin Diseases. J. Investig. Dermatol. 2011, 131, 565–571. [Google Scholar] [CrossRef]
- Li, Q.; Sadowski, S.; Frank, M.; Chai, C.; Varadi, A.; Ho, S.Y.; Lou, H.; Dean, M.; Thisse, C.; Thisse, B.; et al. The abcc6a gene expression is required for normal zebrafish development. J. Investig. Dermatol. 2010, 130, 2561–2568. [Google Scholar] [CrossRef]
- Li, Q.; Guo, H.; Chou, D.W.; Berndt, A.; Sundberg, J.P.; Uitto, J.; Sato, M. Mouse Models for Pseudoxanthoma Elasticum: Genetic and Dietary Modulation of the Ectopic Mineralization Phenotypes. PLoS ONE 2014, 9, e89268. [Google Scholar] [CrossRef]
- Gorgels, T.G.; Hu, X.; Scheffer, G.L.; van der Wal, A.C.; Toonstra, J.; de Jong, P.T.; van Kuppevelt, T.H.; Levelt, C.N.; de Wolf, A.; Loves, W.J.; et al. Disruption of Abcc6 in the mouse: Novel insight in the pathogenesis of pseudoxanthoma elasticum. Hum. Mol. Genet. 2005, 14, 1763–1773. [Google Scholar] [CrossRef]
- Klement, J.F.; Matsuzaki, Y.; Jiang, Q.-J.; Terlizzi, J.; Choi, H.Y.; Fujimoto, N.; Li, K.; Pulkkinen, L.; Birk, D.E.; Sundberg, J.P.; et al. Targeted Ablation of the Abcc6 Gene Results in Ectopic Mineralization of Connective Tissues. Mol. Cell. Biol. 2005, 25, 8299–8310. [Google Scholar] [CrossRef]
- Heston, W.E.; Vlahakis, G. Mammary tumors, plaques, and hyperplastic alveolar nodules in various combinations of mouse inbred strains and the different lines of the mammary tumor virus. Int. J. Cancer 1971, 7, 141–148. [Google Scholar] [CrossRef]
- Aherrahrou, Z.; Doehring, L.C.; Ehlers, E.-M.; Liptau, H.; Depping, R.; Linsel-Nitschke, P.; Kaczmarek, P.M.; Erdmann, J.; Schunkert, H. An Alternative Splice Variant in Abcc6, the Gene Causing Dystrophic Calcification, Leads to Protein Deficiency in C3H/He Mice. J. Biol. Chem. 2008, 283, 7608–7615. [Google Scholar] [CrossRef]
- Kauffenstein, G.; Pizard, A.; Le Corre, Y.; Vessières, E.; Grimaud, L.; Toutain, B.; Labat, C.; Mauras, Y.; Gorgels, T.; Bergen, A.; et al. Disseminated Arterial Calcification and Enhanced Myogenic Response Are Associated With Abcc6 Deficiency in a Mouse Model of Pseudoxanthoma Elasticum. Arter. Thromb. Vasc. Biol. 2014, 34, 1045–1056. [Google Scholar] [CrossRef]
- Hosen, M.J.; Coucke, P.J.; Le Saux, O.; De Paepe, A.; Vanakker, O.M. Perturbation of specific pro-mineralizing signalling pathways in human and murine pseudoxanthoma elasticum. Orphanet J. Rare Dis. 2014, 9, 66. [Google Scholar] [CrossRef]
- Szeri, F.; Miko, A.; Navasiolava, N.; Kaposi, A.; Verschuere, S.; Molnar, B.; Li, Q.; Terry, S.F.; Boraldi, F.; Uitto, J.; et al. The pathogenic c.1171A>G (p.Arg391Gly) and c.2359G>A (p.Val787Ile) ABCC6 variants display incomplete penetrance causing pseudoxanthoma elasticum in a subset of individuals. Hum. Mutat. 2022, 43, 1872–1881. [Google Scholar] [CrossRef] [PubMed]
- Köblös, G.; Andrikovics, H.; Prohászka, Z.; Tordai, A.; Váradi, A.; Arányi, T. The R1141X Loss-of-Function Mutation of the ABCC6 Gene Is a Strong Genetic Risk Factor for Coronary Artery Disease. Genet. Test. Mol. Biomark. 2010, 14, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, L.; Tarugi, P.; Borrini, C.; Bellocchio, A.; Fresa, R.; Guerra, D.; Quaglino, D.; Ronchetti, I.; Calandra, S.; Bertolini, S. Pseudoxanthoma elasticum and familial hypercholes-terolemia: A deleterious combination of cardiovascular risk factors. Atherosclerosis 2010, 210, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Pseudoxanthoma elasticum. Orphanet J. Rare Dis. 2017, 12, 85. [Google Scholar] [CrossRef]
- Sato, N.; Nakayama, T.; Mizutani, Y.; Yuzawa, M. Novel mutations of ABCC6 gene in Japanese patients with Angioid streaks. Biochem. Biophys. Res. Commun. 2009, 380, 548–553. [Google Scholar] [CrossRef]
- Li, Q.; Sadowski, S.; Uitto, J. Angioid Streaks in Pseudoxanthoma Elasticum: Role of the p.R1268Q Mutation in the ABCC6 Gene. J. Investig. Dermatol. 2011, 131, 782–785. [Google Scholar] [CrossRef]
- Omarjee, L.; Nitschke, Y.; Verschuere, S.; Bourrat, E.; Vignon, M.D.; Navasiolava, N.; Leftheriotis, G.; Kauffenstein, G.; Rutsch, F.; Vanakker, O.M.; et al. Severe early-onset manifestations of pseudoxanthoma elasticum resulting from the cumulative effects of several deleterious mutations in ENPP1, ABCC6 and HBB: Transient improvement in ectopic calcification with sodium thiosulfate. Br. J. Dermatol. 2020, 183, 367–372. [Google Scholar] [CrossRef]
- Kalal, I.G.; Seetha, D.; Panda, A.; Nitschke, Y.; Rutsch, F. Molecular diagnosis of generalized arterial calcification of infancy (GACI). J. Cardiovasc. Dis. Res. 2012, 3, 150–154. [Google Scholar] [CrossRef]
- Lebwohl, M.; Neldner, K.; Pope, F.M.; De Paepe, A.; Christiano, A.M.; Boyd, C.D.; Uitto, J.; McKusick, V.A. Classification of pseudoxanthoma elasticum: Report of a consensus conference. J. Am. Acad. Dermatol. 1994, 30, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Bercovitch, L.; Terry, S.F.; Terry, P.F. Pseudoxanthoma elasticum: Progress in diagnostics and research towards treatment: Summary of the 2010 PXE International Research Meeting. Am. J. Med. Genet. A 2011, 155, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Plomp, A.S.; Toonstra, J.; Bergen, A.A.; van Dijk, M.R.; de Jong, P.T. Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am. J. Med. Genet. Part. A 2010, 152, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ncbi.nlm.nih.gov/clinvar/?term=ABCC6[gene] (accessed on 18 May 2025).
- Larusso, J.; Ringpfeil, F.; Uitto, J. Pseudoxanthoma elasticum: A streamlined, ethnicity-based mutation detection strategy. Clin. Transl. Sci. 2010, 3, 295–298. [Google Scholar] [CrossRef]
- Jin, L.; Jiang, Q.; Wu, Z.; Shao, C.; Zhou, Y.; Yang, L.; Uitto, J.; Wang, G. Genetic Heterogeneity of Pseudoxanthoma Elasticum: The Chinese Signature Profile of ABCC6 and ENPP1 Mutations. J. Investig. Dermatol. 2015, 135, 1294–1302. [Google Scholar] [CrossRef]
- Pomozi, V.; Brampton, C.; Fülöp, K.; Chen, L.H.; Apana, A.; Li, Q.; Uitto, J.; Le Saux, O.; Váradi, A. Analysis of pseudoxanthoma elasticum-causing missense mutants of ABCC6 in vivo; pharmacological correction of the mislocalized proteins. J. Investig. Dermatol. 2014, 134, 946–953. [Google Scholar] [CrossRef]
- Germain, D.P. Pseudoxanthoma elasticum: Evidence for the existence of a pseudogene highly homologous to the ABCC6 gene. J. Med. Genet. 2001, 38, 457–461. [Google Scholar] [CrossRef]
- Pulkkinen, L.; Nakano, A.; Ringpfeil, F.; Uitto, J. Identification of ABCC6 pseudogenes on human chromosome 16p: Implications for mutation detection in pseudoxanthoma elasticum. Hum. Genet. 2001, 109, 356–365. [Google Scholar] [CrossRef]
- Kringen, M.K.; Stormo, C.; Grimholt, R.M.; Berg, J.P.; Piehler, A.P. Copy number variations of the ATP-binding cassette transporter ABCC6 gene and its pseudogenes. BMC Res. Notes 2012, 5, 425. [Google Scholar] [CrossRef]
- Kringen, M.K.; Stormo, C.; Berg, J.P.; Terry, S.F.; Vocke, C.M.; Rizvi, S.; Hendig, D.; Piehler, A.P. Copy number variation in the ATP-binding cassette transporter ABCC6 gene and ABCC6 pseudogenes in patients with pseudoxanthoma elasticum. Mol. Genet. Genom. Med. 2015, 3, 233–237. [Google Scholar] [CrossRef]
- Prunier, F.; Terrien, G.; Le Corre, Y.; Apana, A.L.Y.; Bière, L.; Kauffenstein, G.; Furber, A.; Bergen, A.A.B.; Gorgels, T.G.M.F.; Le Saux, O.; et al. Pseudoxanthoma Elasticum: Cardiac Findings in Patients and Abcc6-Deficient Mouse Model. PLoS ONE 2013, 8, e68700. [Google Scholar] [CrossRef] [PubMed]
- Hendig, D.; Knabbe, C.; Götting, C. New insights into the pathogenesis of pseudoxanthoma elasticum and related soft tissue calcification disorders by identifying genetic interactions and modifiers. Front. Genet. 2013, 4, 43253. [Google Scholar] [CrossRef]
- Shi, Y.; Terry, S.F.; Terry, P.F.; Bercovitch, L.G.; Gerard, G.F. Development of a Rapid, Reliable Genetic Test for Pseudoxanthoma Elasticum. J. Mol. Diagn. 2007, 9, 105–112. [Google Scholar] [CrossRef]
- Legrand, A.; Cornez, L.; Samkari, W.; Mazzella, J.-M.; Venisse, A.; Boccio, V.; Auribault, K.; Keren, B.; Benistan, K.; Germain, D.P.; et al. Mutation spectrum in the ABCC6 gene and genotype–phenotype correlations in a French cohort with pseudoxanthoma elasticum. Anesth. Analg. 2017, 19, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; De Zaeytijd, J.; Voet, D.; De Paepe, A.; De Backer, T.; De Backer, J. Characterization of Cardiovascular Involvement in Pseudoxanthoma Elasticum Families. Arter. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, C.; Dichgans, M. Small vessel disease: Mechanisms and clinical implications. Lancet Neurol. 2019, 18, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Duering, M.; Biessels, G.J.; Brodtmann, A.; Chen, C.; Cordonnier, C.; de Leeuw, F.-E.; Debette, S.; Frayne, R.; Jouvent, E.; Rost, N.S.; et al. Neuroimaging standards for research into small vessel disease—Advances since 2013. Lancet Neurol. 2023, 22, 602–618. [Google Scholar] [CrossRef]
- O’Brien, J.T.; Thomas, A. Vascular dementia. Lancet 2015, 386, 1698–1706. [Google Scholar] [CrossRef]
- Debette, S.; Schilling, S.; Duperron, M.-G.; Larsson, S.C.; Markus, H.S. Clinical Significance of Magnetic Resonance Imaging Markers of Vascular Brain Injury. JAMA Neurol. 2019, 76, 81–94. [Google Scholar] [CrossRef]
- Tan, R.; Traylor, M.; Rutten-Jacobs, L.; Markus, H. New insights into mechanisms of small vessel disease stroke from genetics. Clin. Sci. 2017, 131, 515–531. [Google Scholar] [CrossRef]
- Uemura, M.; Nozaki, H.; Kato, T.; Koyama, A.; Sakai, N.; Ando, S.; Kanazawa, M.; Hishikawa, N.; Nishimoto, Y.; Polavarapu, K.; et al. HTRA1-Related Cerebral Small Vessel Disease: A Review of the Literature. Front. Neurol. 2020, 11, 545. [Google Scholar] [CrossRef] [PubMed]
- Chabriat, H.; Joutel, A.; Dichgans, M.; Tournier-Lasserve, E.; Bousser, M.-G. CADASIL. Lancet Neurol. 2009, 8, 643–653. [Google Scholar] [CrossRef]
- Tan, R.Y.Y.; Markus, H.S. Monogenic causes of stroke: Now and the future. J. Neurol. 2015, 262, 2601–2616. [Google Scholar] [CrossRef]
- Tan, R.Y.; Traylor, M.; Megy, K.; Duarte, D.; Deevi, S.V.; Shamardina, O.; Mapeta, R.P.; Ouwehand, W.H.; Gräf, S.; Downes, K.; et al. How common are single gene mutations as a cause for lacunar stroke? Neurology 2019, 93, e2007–e2020. [Google Scholar] [CrossRef]
- Kilarski, L.L.; Rutten-Jacobs, L.C.; Bevan, S.; Baker, R.; Hassan, A.; Hughes, D.A.; Markus, H.S.; UK Young Lacunar Stroke DNA Study. Prevalence of CADASIL and Fabry disease in a cohort of MRI defined younger onset lacunar stroke. PLoS ONE 2015, 10, e0136352. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.P.H.; Auckland, K.; Gräf, S.; Markus, H.S. Rare Sequence Variation Underlying Suspected Familial Cerebral Small-Vessel Disease. J. Am. Hear. Assoc. 2024, 13, e035771. [Google Scholar] [CrossRef]
- Uemura, M.; Hatano, Y.; Nozaki, H.; Ando, S.; Kondo, H.; Hanazono, A.; Iwanaga, A.; Murota, H.; Osakada, Y.; Osaki, M.; et al. High frequency of HTRA1 and ABCC6 mutations in Japanese patients with adult-onset cerebral small vessel disease. J. Neurol. Neurosurg. Psychiatry 2022, 94, 74–81. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.; Cardoen, S.; Hosen, M.J.; Le Saux, O.; De Zaeytijd, J.; Leroy, B.P.; De Reuck, J.; Coucke, P.J.; De Paepe, A.; Hemelsoet, D.; et al. Pathogenic variants in the ABCC6 gene are associated with an increased risk for ischemic stroke. Brain Pathol. 2018, 28, 822–831. [Google Scholar] [CrossRef]
- Li, C.; Issa, R.; Kumar, P.; Hampson, I.N.; Lopez-Novoa, J.M.; Bernabeu, C.; Kumar, S. CD105 prevents apoptosis in hypoxic endothelial cells. J. Cell. Sci. 2003, 116, 2677–2685. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Zhao, P.; Yao, Y.; Meng, H.; Rau, C.; Havel, J.V.; Gorgels, T.G.; Bergen, A.A.; MacLellan, W.R.; Drake, T.A.; et al. Abcc6 deficiency causes increased infarct size and apoptosis in a mouse cardiac ischemia-reperfusion model. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2806–2812. [Google Scholar] [CrossRef]
- Rajagopal, R.; Dattilo, L.K.; Kaartinen, V.; Deng, C.X.; Umans, L.; Zwijsen, A.; Roberts, A.B.; Bottinger, E.P.; Beebe, D.C. Functions of the type 1 BMP receptor Acvr1 (Alk2) in lens development: Cell proliferation, terminal differentiation, and survival. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Gheduzzi, D.; Boraldi, F.; Annovi, G.; DeVincenzi, C.P.; Schurgers, L.J.; Vermeer, C.; Quaglino, D.; Ronchetti, I.P. Matrix Gla protein is involved in elastic fiber calcification in the dermis of pseudoxanthoma elasticum patients. Mod. Pathol. 2007, 87, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Kauw, F.; Kranenburg, G.; Kappelle, L.J.; Hendrikse, J.; Koek, H.L.; Visseren, F.L.J.; Mali, W.P.T.; de Jong, P.A.; Spiering, W. Cerebral disease in a nationwide Dutch pseudoxanthoma elasticum cohort with a systematic review of the literature. J. Neurol. Sci. 2017, 15, 167–172. [Google Scholar] [CrossRef]
- Andrikovics, H.; Pongrácz, E.; Kalina, E.; Szilvási, A.; Aslanidis, C.; Schmitz, G.; Tordai, A. Decreased frequencies of ABCA1 polymorphisms R219K and V771M in Hungarian patients with cerebrovascular and cardiovascular diseases. Cerebrovasc. Dis. 2006, 21, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Sisodiya, S.M.; Lin, W.; Harding, B.N.; Squier, M.V.; Thom, M. Drug resistance in epilepsy: Expression of drug resistance proteins in common causes of refractory epilepsy. Brain 2002, 125, 22–31. [Google Scholar] [CrossRef]
- Spudich, A.; Kilic, E.; Xing, H.; Kilic, U.; Rentsch, K.M.; Wunderli-Allenspach, H.; Bassetti, C.L.; Hermann, D.M. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat. Neurosci. 2006, 9, 487–488. [Google Scholar] [CrossRef]
- Pachori, A.S.; Custer, L.; Hansen, D.; Clapp, S.; Kemppa, E.; Klingensmith, J. Bone morphogenetic protein 4 mediates myocardial ischemic injury through JNK-dependent signaling pathway. J. Mol. Cell. Cardiol. 2010, 48, 1255–1265. [Google Scholar] [CrossRef]
- Pacifici, M.; Shore, E.M. Common mutations in ALK2/ACVR1, a multi-faceted receptor, have roles in distinct pediatric musculoskeletal and neural orphan disorders. Cytokine Growth Factor Rev. 2015, 27, 93–104. [Google Scholar] [CrossRef]
- Zhu, Y.; Sun, Y.; Xie, L.; Jin, K.; Sheibani, N.; Greenberg, D.A. Hypoxic Induction of Endoglin via Mitogen-Activated Protein Kinases in Mouse Brain Microvascular Endothelial Cells. Stroke 2003, 34, 2483–2488. [Google Scholar] [CrossRef]
- Brampton, C.; Pomozi, V.; Chen, L.-H.; Apana, A.; McCurdy, S.; Zoll, J.; Boisvert, W.A.; Lambert, G.; Henrion, D.; Blanchard, S.; et al. ABCC6 deficiency promotes dyslipidemia and atherosclerosis. Sci. Rep. 2021, 11, 3881. [Google Scholar] [CrossRef]
- Hu, X.; Peek, R.; Plomp, A.; Brink, J.T.; Scheffer, G.; van Soest, S.; Leys, A.; de Jong, P.T.V.M.; Bergen, A.A.B. Analysis of the Frequent R1141X Mutation in the ABCC6 Gene in Pseudoxanthoma Elasticum. Investig. Opthalmology Vis. Sci. 2003, 44, 1824–1829. [Google Scholar] [CrossRef] [PubMed]
- Kuzaj, P.; Kuhn, J.; Dabisch-Ruthe, M.; Faust, I.; Götting, C.; Knabbe, C.; Hendig, D. ABCC6- a new player in cellular cholesterol and lipoprotein metabolism? Lipids Health Dis. 2014, 13, 118. [Google Scholar] [CrossRef]
- Wang, J.; Near, S.; Young, K.; Connelly, P.W.; Hegele, R.A. ABCC6 gene polymorphism associated with variation in plasma lipoproteins. J. Hum. Genet. 2001, 46, 699–705. [Google Scholar] [CrossRef]
- Miglionico, R.; Armentano, M.F.; Carmosino, M.; Salvia, A.M.; Cuviello, F.; Bisaccia, F.; Ostuni, A. Dysregulation of gene expression in ABCC6 knockdown HepG2 cells. Cell. Mol. Biol. Lett. 2014, 19, 517–526. [Google Scholar] [CrossRef]
- Stumpf, M.J.; Mahn, T.; Steinmetz, M.; Fimmers, R.; Pizarro, C.; Nickenig, G.; Skowasch, D.; Schahab, N.; Schaefer, C.A. Pseudoxanthoma elasticum—Also a microvascular disease. Vasa 2020, 49, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Miglionico, R.; Ostuni, A.; Armentano, M.F.; Milella, L.; Crescenzi, E.; Carmosino, M.; Bisaccia, F. ABCC6 knockdown in HepG2 cells induces a senescent-like cell phenotype. Cell. Mol. Biol. Lett. 2017, 22, 7. [Google Scholar] [CrossRef]
- Wang, S.S.; Martin, L.J.; Schadt, E.E.; Meng, H.; Wang, X.; Zhao, W.; Ingram-Drake, L.; Nebohacova, M.; Mehrabian, M.; Drake, T.A.; et al. Disruption of the Aortic Elastic Lamina and Medial Calcification Share Genetic Determinants in Mice. Circ. Cardiovasc. Genet. 2009, 2, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Bartstra, J.W.; van den Beukel, T.; Kranenburg, G.; Geurts, L.J.; den Harder, A.M.; Witkamp, T.; Wolterink, J.M.; Zwanenburg, J.J.M.; van Valen, E.; Koek, H.L.; et al. Increased Intracranial Arterial Pulsatility and Micro-vascular Brain Damage in Pseudoxanthoma Elasticum. AJNR Am. J. Neuroradiol. 2024, 45, 386–392. [Google Scholar] [CrossRef]
- Kranenburg, G.; De Jong, P.A.; Mali, W.P.; Attrach, M.; Visseren, F.L.J.; Spiering, W. Prevalence and severity of arterial calcifications in pseudoxanthoma elasticum (PXE) compared to hospital controls. Novel insights into the vascular phenotype of PXE. Atherosclerosis 2017, 256, 7–14. [Google Scholar] [CrossRef]
- van Tuijl, R.J.; Ruigrok, Y.M.; Geurts, L.J.; van der Schaaf, I.C.; Biessels, G.J.; Rinkel, G.J.E.; Velthuis, B.K.; Zwanenburg, J.J.M. Does the Internal Carotid Artery Attenuate Blood-Flow Pulsatility in Small Vessel Disease? A 7 T 4D-Flow MRI Study. J. Magn. Reson. Imaging 2022, 56, 527–535. [Google Scholar] [CrossRef]
- Risseeuw, S.; Ossewaarde-van Norel, J.; van Buchem, C.; Spiering, W.; Imhof, S.M.; van Leeuwen, R. The Extent of Angioid Streaks Correlates With Macular Degeneration in Pseudoxanthoma Elasticum. Am. J. Ophthalmol. 2020, 220, 82–90. [Google Scholar] [CrossRef]
- Mitchell, G.F. Aortic stiffness, pressure and flow pulsatility, and target organ damage. J. Appl. Physiol. 2018, 125, 1871–1880. [Google Scholar] [CrossRef] [PubMed]
- Zedde, M.; Pascarella, R. Arterial Calcification as a Pseudoxanthoma Elasticum-like Manifestation in Beta-Thalassemia: Molecular Mechanisms and Significance. Hemato 2025, 6, 7. [Google Scholar] [CrossRef]
- Leftheriotis, G.; Kauffenstein, G.; Hamel, J.F.; Abraham, P.; Le Saux, O.; Willoteaux, S.; Henrion, D.; Martin, L.; Eller, P. The Contribution of Arterial Calcification to Peripheral Arterial Disease in Pseudoxanthoma Elasticum. PLoS ONE 2014, 9, e96003. [Google Scholar] [CrossRef] [PubMed]
- Bartstra, J.W.; Spiering, W.; Ouweland, J.M.W.v.D.; Mali, W.P.T.M.; Janssen, R.; de Jong, P.A. Increased Elastin Degradation in Pseudoxanthoma Elasticum Is Associated with Peripheral Arterial Disease Independent of Calcification. J. Clin. Med. 2020, 9, 2771. [Google Scholar] [CrossRef]
- Tsao, C.W.; Pencina, K.M.; Massaro, J.M.; Benjamin, E.J.; Levy, D.; Vasan, R.S.; Hoffmann, U.; O’Donnell, C.J.; Mitchell, G.F. Cross-Sectional Relations of Arterial Stiffness, Pressure Pulsatility, Wave Reflection, and Arterial Calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2495–2500. [Google Scholar] [CrossRef] [PubMed]
- Hong, N.R.; Seo, H.S.; Lee, Y.H.; Kim, J.H.; Seol, H.Y.; Lee, N.J.; Suh, S.-I. The correlation between carotid siphon calcification and lacunar infarction. Neuroradiology 2011, 53, 643–649. [Google Scholar] [CrossRef]
- Melgarejo, J.D.; Vernooij, M.W.; Ikram, M.A.; Zhang, Z.-Y.; Bos, D. Intracranial Carotid Arteriosclerosis Mediates the Association Between Blood Pressure and Cerebral Small Vessel Disease. Hypertension 2023, 80, 618–628. [Google Scholar] [CrossRef]
- Omarjee, L.; Fortrat, J.-O.; Larralde, A.; Le Pabic, E.; Kauffenstein, G.; Laot, M.; Navasiolava, N.; Mention, P.-J.; Linares, J.L.C.; Valdivielso, P.; et al. Internal Carotid Artery Hypoplasia: A New Clinical Feature in Pseudoxanthoma Elasticum. J. Stroke 2019, 21, 108–111. [Google Scholar] [CrossRef]
- Mention, P.-J.; Lacoeuille, F.; Leftheriotis, G.; Martin, L.; Omarjee, L. 18F-Flurodeoxyglucose and 18F-Sodium Fluoride Positron Emission Tomography/Computed Tomography Imaging of Arterial and Cutaneous Alterations in Pseudoxanthoma Elasticum. Circ. Cardiovasc. Imaging 2018, 11, e007060. [Google Scholar] [CrossRef]
- Tanase, D.M.; Valasciuc, E.; Anton, I.-B.; Gosav, E.M.; Dima, N.; Cucu, A.I.; Costea, C.F.; Floria, D.E.; Hurjui, L.L.; Tarniceriu, C.C.; et al. Matrix Metalloproteinases: Pathophysiologic Implications and Potential Therapeutic Targets in Cardiovascular Disease. Biomolecules 2025, 15, 598. [Google Scholar] [CrossRef] [PubMed]
- Wolosowicz, M.; Prokopiuk, S.; Kaminski, T.W. The Complex Role of Matrix Metalloproteinase-2 (MMP-2) in Health and Disease. Int. J. Mol. Sci. 2024, 25, 13691. [Google Scholar] [CrossRef] [PubMed]
- Di Nubila, A.; Dilella, G.; Simone, R.; Barbieri, S.S. Vascular Extracellular Matrix in Atherosclerosis. Int. J. Mol. Sci. 2024, 25, 12017. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, C.; Galaction, A.I.; Poștaru, M.; Rotariu, M.; Turnea, M.; Blendea, C.D. Hydrogen Sulfide Modulation of Matrix Metalloproteinases and CD147/EMMPRIN: Mechanistic Pathways and Impact on Atherosclerosis Progression. Biomedicines 2024, 12, 1951. [Google Scholar] [CrossRef] [PubMed]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharmacol. 2008, 75, 346–359. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Molecular Mechanism | Description |
|---|---|
| ABCC6 function loss | Decreased functionality of ABCC6 affects the transport of critical substrates, leading to metabolic disruptions. |
| ATP secretion | ABCC6-dependent ATP secretion from the liver is impaired; ATP is not directly transported by ABCC6. |
| Low Pyrophosphate (PPi) Levels | ABCC6 deficiency results in low circulating PPi levels, which are crucial for preventing ectopic mineralization. |
| Increased Inorganic Phosphate (Pi) | Elevated Pi levels may promote calcification; the low PPi/Pi ratio is implicated in PXE’s pathophysiology. |
| Ectopic Mineralization | Dysfunctional ABCC6 leads to mineralization in soft tissues (skin, eyes, arteries) due to the lack of inhibitory factors. |
| Altered Expression of Anti-mineralization Proteins | Impaired export of proteins like matrix Gla-protein (MGP) and fetuin-A, which normally inhibit mineralization. |
| Oxidative Stress | Increased oxidative stress may contribute to cellular damage and exacerbate PXE manifestations. |
| Dysregulation of Signaling Pathways | Activation of pathways such as BMP2-SMAD-RUNX2 associated with vascular calcification in the absence of ABCC6. |
| Major Diagnostic Criteria | |
|---|---|
| 1. Skin | |
| a. | Yellowish papules and/or plaques on the lateral side of the neck and/or flexural areas of the body; or |
| b. | Increase in morphologically altered elastin with fragmentation, clumping, and calcification of elastic fibers in a skin biopsy from clinically affected skin. |
| 2. Eye | |
| a. | Peau d’orange of the retina; or |
| b. | One or more angioid streaks (ASs), each at least as long as one disk diameter. If uncertain, fluorescein or indocyanine green angiography of the fundus is necessary for confirmation. |
| 3. Genetics | |
| a. | A pathogenic mutation of both alleles of the ABCC6 gene; or |
| b. | A first-degree relative (parent, sibling, child) who independently meets the diagnostic criteria for definitive PXE. |
| Minor Diagnostic Criteria | |
| 1. Eye | |
| a. | One angioid streak shorter than one disk diameter; or |
| b. | One or more ‘comets’ in the retina; or |
| c. | One or more ‘wing signs’ in the retina. |
| 2. Genetics | |
| a. | A pathogenic mutation of one allele of the ABCC6 gene. |
| Requirements for the Diagnosis of PXE | |
| a. Definitive Diagnosis | Presence of two (or more) major criteria not belonging to the same (skin, eye, genetic) category. |
| b. Probable Diagnosis | Presence of two major eye criteria or two major skin criteria, or one major criterion and one or more minor criteria not belonging to the same category as the major criterion. |
| c. Possible Diagnosis | Presence of a single major criterion, or one or more minor criteria. |
| Mechanism | Description |
|---|---|
| Dysfunction of BMP and TGFβ Signaling | Dysregulation of bone morphogenetic protein (BMP) and transforming growth factor β (TGFβ) pathways in ABCC6-deficient models suggests a pro-ischemic state. |
| Pro-apoptotic Factors | Upregulation of Bmp4 and downregulation of Alk2 observed in brain tissue of Abcc6-deficient mice, leading to increased apoptosis. |
| Increased Cardiovascular Risk | Pathogenic ABCC6 variants correlate with increased cardiovascular risk; the mechanisms may involve other ABC transporters. |
| Dyslipidemia and Atherosclerosis | ABCC6 deficiency linked to altered lipoprotein profiles, decreased HDL, and increased LDL, contributing to atherosclerosis. |
| Systemic Inflammation | Increased pro-inflammatory cytokines (e.g., IL-6, CCL-2) in ABCC6-deficient models, exacerbating atherosclerotic phenotype. |
| Lipid Metabolism | ABCC6 deficiency modulates plasma lipoproteins; significant reductions in HDL levels observed in PXE patients. |
| Indirect Pathways | ABCC6 deficiency may contribute to classic vascular risk factors, influencing the development of ischemic strokes. |
| Technique | Details |
|---|---|
| 3T MRI Protocol | Patients underwent a standardized MR imaging protocol using a 3T MRI scanner. This high-field strength allows for better resolution and detail in imaging.
|
| CT |
|
| Image Processing | Custom MATLAB scripts were employed to analyze the 2D phase-contrast acquisitions, creating regions of interest (ROIs) for blood flow measurements and correcting potential phase wraps in the velocity maps. |
| Segmentation | Automatic segmentation of brain volumes and WML was performed using the Computational Anatomy Toolbox and the Lesion Segmentation Tool within SPM12 software. This automated approach allowed for the precise quantification of brain structures and lesions. |
| Biomarkers | Description |
|---|---|
| Matrix Metalloproteinases (MMPs) |
|
| Tissue Inhibitors of Metalloproteinases (TIMPs) |
|
| Collagen Degradation Products |
|
| Elastin Degradation Products | Desmosine and Isodesmosine: Markers of elastin degradation, associated with vascular elasticity and stiffness. |
| Fibronectin and its Fragments | Elevated levels can indicate matrix remodeling and are associated with cardiovascular risk |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zedde, M.; Pascarella, R. ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations. Genes 2025, 16, 728. https://doi.org/10.3390/genes16070728
Zedde M, Pascarella R. ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations. Genes. 2025; 16(7):728. https://doi.org/10.3390/genes16070728
Chicago/Turabian StyleZedde, Marialuisa, and Rosario Pascarella. 2025. "ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations" Genes 16, no. 7: 728. https://doi.org/10.3390/genes16070728
APA StyleZedde, M., & Pascarella, R. (2025). ABCC6 Involvement in Cerebral Small Vessel Disease: Potential Mechanisms and Associations. Genes, 16(7), 728. https://doi.org/10.3390/genes16070728