
Genomic Regions Associated with Respiratory Disease in Holstein Calves in the Southern United States



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genotyping
2.3. Quality Control
2.4. Genome-Wide Association Analysis
2.5. Positional Candidate Genes
2.6. Gene Set Enrichment Analysis–Single Nucleotide Polymorphism
3. Results
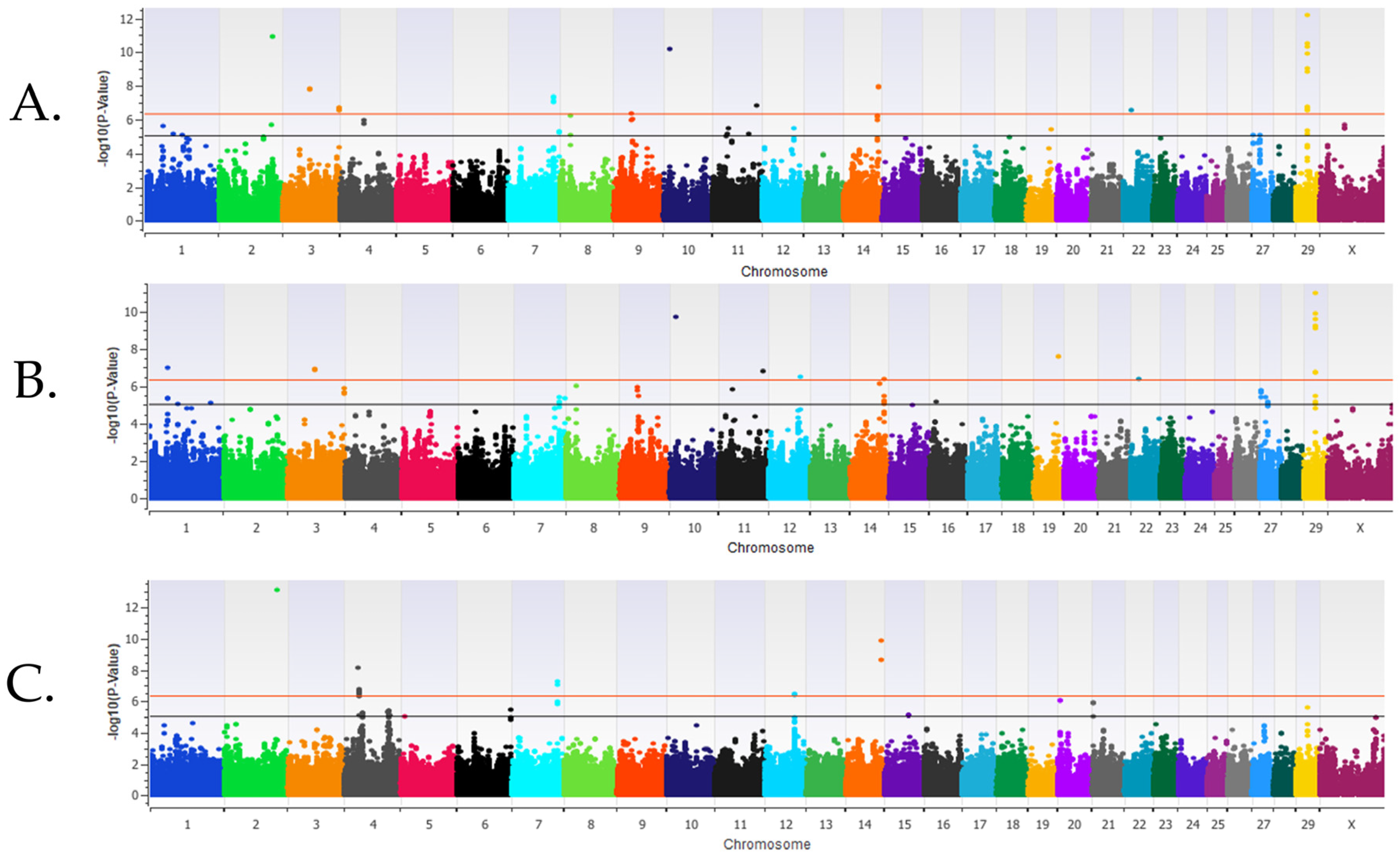
3.1. Genome-Wide Association Analysis Results
3.2. Gene Set Enrichment Analysis with Single Nucleotide Polymorphisms
3.3. Shared Genes and Gene Sets
4. Discussion
4.1. Genome-Wide Association Analysis of Pre-Weaned Heifers
4.2. Gene Set Enrichment Analysis—Single Nucleotide Polymorphisms of Pre-Weaned Heifers
4.3. Genome-Wide Association Analysis of Post-Weaned Heifers
4.4. Gene Set Enrichment Analysis—Single Nucleotide Polymorphisms in Post-Weaned Heifers
4.5. Sharing of Bovine Respiratory Disease Positional Candidate and Leading-Edge Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaffer, A.P.; Larson, R.L.; Cernicchiaro, N.; Hanzlicek, G.A.; Bartle, S.J.; Thomson, D.U. The association between calfhood bovine respiratory disease complex and subsequent departure from the herd, milk production, and reproduction in dairy cattle. J. Am. Vet. Med. Assoc. 2016, 248, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- National Animal Health Monitoring System. United States Department of Agriculture. Health and Management Practices on U.S. Dairy Operations, 2014. Report 3. 2018. Available online: https://www.aphis.usda.gov/sites/default/files/dairy14_dr_partiii.pdf (accessed on 21 April 2025).
- Urie, N.J.; Lombard, J.E.; Shivley, C.B.; Kopral, C.A.; Adams, A.E.; Earleywine, T.J.; Olson, J.D.; Garry, F.B. Preweaned heifer management on US dairy operations: Part V. factors associated with morbidity and mortality in preweaned dairy heifer calves. J. Dairy Sci. 2018, 101, 9229–9244. [Google Scholar] [CrossRef] [PubMed]
- Klima, C.L.; Zaheer, R.; Cook, S.R.; Booker, C.W.; Hendrick, S.; Alexander, T.W.; McAllister, T.A. Pathogens of bovine respiratory disease in North American feedlots conferring multidrug resistance via integrative conjugative elements. J. Clin. Microbiol. 2014, 52, 438–448. [Google Scholar] [CrossRef] [PubMed]
- McGill, J.L.; Sacco, R.E. The immunology of bovine respiratory disease. Vet. Clin. North. Am. Food. Anim. Pract. 2020, 36, 333–348. [Google Scholar] [CrossRef]
- Gulliksen, S.M.; Lie, K.I.; Løken, T.; Østerås, O. Calf mortality in Norwegian dairy herds. J. Dairy Sci. 2009, 92, 2782–2795. [Google Scholar] [CrossRef]
- Lago, A.; McGuirk, S.M.; Bennett, T.B.; Cook, N.B.; Nordlund, K.V. Calf respiratory disease and pen microenvironments in naturally ventilated calf barns in winter. J. Dairy Sci. 2006, 89, 4014–4025. [Google Scholar] [CrossRef]
- Berry, D.P.; Bermingham, M.L.; Good, M.; More, S.J. Genetics of Animal Health and disease in cattle. Ir. Vet. J. 2011, 64, 1–10. [Google Scholar] [CrossRef]
- Hayes, B.J.; Duff, C.J.; Hine, B.C.; Mahony, T.J. Genomic estimated breeding values for bovine respiratory disease resistance in Angus Feedlot Cattle. J. Anim. Sci. 2024, 102, skae113. [Google Scholar] [CrossRef]
- Kiser, J.N.; Lawrence, T.E.; Neupane, M.; Seabury, C.M.; Neibergs, H.L. Rapid communication: Subclinical bovine respiratory disease—Loci and pathogens associated with lung lesions in feedlot cattle. J. Anim. Sci. 2017, 95, 2726–2731. [Google Scholar] [CrossRef]
- Neibergs, H.L.; Seabury, C.M.; Wojtowicz, A.J.; Wang, Z.; Scraggs, E.; Kiser, J.N.; Neupane, M.; Womack, J.E.; VanEenennaam, A.L.; Hagevoort, G.R.; et al. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned Holstein calves. BMC Genomics 2014, 15, 1164. [Google Scholar] [CrossRef]
- VanRaden, P.M.; Cole, J.B.; Neupane, M.; Toghiani, S.; Gaddis, K.L.; Tempelman, R.J. Net Merit as a Measure of Lifetime Profit: 2021 Revision. USDA-ARS-AGIL, Beltsville, MD 20705-2350, NM$8 (05-21). 2021. Available online: https://www.ars.usda.gov/ARSUserFiles/80420530/Publications/ARR/nmcalc-2021_ARR-NM8.pdf (accessed on 18 March 2025).
- Zoetis. Dairy Wellness Profit Index® Enhancements for Profitable Genetic Selection and Helping Sustainable Practices. 2022. Available online: https://www.zoetisus.com/content/pages/Solutions/Dairy/Dairy-Genetics/Dairy-genetics-resources/Documents/Genetics-edetailer.pdf (accessed on 7 April 2025).
- Gonzalez-Peña, D.; Vukasinovic, N.; Brooker, J.J.; Przybyla, C.A.; DeNise, S.K. Genomic evaluation for calf wellness traits in Holstein cattle. J. Dairy Sci. 2019, 102, 2319–2329. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Browning, S.R. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016, 98, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.N.; Keuter, E.M.; Seabury, C.M.; Neupane, M.; Moraes, J.G.; Dalton, J.; Burns, G.W.; Spencer, T.E.; Neibergs, H.L. Validation of 46 loci associated with female fertility traits in cattle. BMC Genomics 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kiser, J.N.; Clancey, E.; Moraes, J.G.; Dalton, J.; Burns, G.W.; Spencer, T.E.; Neibergs, H.L. Identification of loci associated with conception rate in primiparous Holstein Cows. BMC Genomics 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Devlin, B.; Roeder, K. Genomic Control for Association Studies. Biometrics 1999, 55, 997–1004. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Weiss, K.M.; Clark, A.G. Linkage disequilibrium and the mapping of complex human traits. Trends Genet. 2002, 18, 19–24. [Google Scholar] [CrossRef]
- Lewontin, R.C. On measures of gametic disequilibrium. Genetics 1988, 120, 849–852. [Google Scholar] [CrossRef]
- Taylor, J.F. Implementation and accuracy of Genomic Selection. Aquaculture 2014, 420–421, S8–S14. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.B.; Schaffner, S.F.; Nguyen, H.; Moore, J.M.; Roy, J.; Blumenstiel, B.; Higgins, J.; DeFelice, M.; Lochner, A.; Faggart, M.; et al. The structure of haplotype blocks in the human genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010, 11, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Karssen, L.C.; van Duijn, C.M.; Aulchenko, Y.S. The GENABEL Project for Statistical Genomics. F1000Res 2016, 5, 914. [Google Scholar] [CrossRef]
- Aulchenko, Y.S.; Ripke, S.; Isaacs, A.; van Duijn, C.M. Genabel: An R library for genome-wide association analysis. Bioinformatics 2007, 23, 1294–1296. [Google Scholar] [CrossRef]
- Neupane, M.; Kiser, J.N.; Neibergs, H.L. Gene set enrichment analysis of snp data in dairy and beef cattle with bovine respiratory disease. Anim. Genet. 2018, 49, 527–538. [Google Scholar] [CrossRef]
- Hasankhani, A.; Bahrami, A.; Sheybani, N.; Fatehi, F.; Abadeh, R.; Ghaem Maghami Farahani, H.; Bahreini Behzadi, M.R.; Javanmard, G.; Isapour, S.; Khadem, H.; et al. Integrated network analysis to identify key modules and potential hub genes involved in bovine respiratory disease: A systems biology approach. Front. Genet. 2021, 12, 753839. [Google Scholar] [CrossRef]
- Strillacci, M.G.; Ferrulli, V.; Bernini, F.; Pravettoni, D.; Bagnato, A.; Martucci, I.; Boccardo, A. Genomic analysis of bovine respiratory disease resistance in preweaned dairy calves diagnosed by a combination of clinical signs and thoracic ultrasonography. PLoS ONE 2025, 20, 318520. [Google Scholar] [CrossRef]
- Cao, H.; Fang, C.; Liu, L.L.; Farnir, F.; Liu, W.J. Identification of Susceptibility Genes Underlying Bovine Respiratory Disease in Xinjiang Brown Cattle Based on DNA Methylation. Int. J. Mol. Sci. 2024, 25, 4928. [Google Scholar] [CrossRef]
- Quick, A.E.; Ollivett, T.L.; Kirkpatrick, B.W.; Weigel, K.A. Genomic analysis of bovine respiratory disease and lung consolidation in preweaned Holstein calves using clinical scoring and lung ultrasound. J. Dairy Sci. 2020, 103, 1632–1641. [Google Scholar] [CrossRef]
- Tizioto, P.C.; Kim, J.; Seabury, C.M.; Schnabel, R.D.; Gershwin, L.J.; Van Eenennaam, A.L.; Toaff-Rosenstein, R.; Neibergs, H.L.; Bovine Respiratory Disease Complex Coordinated Agricultural Project Research Team; Taylor, J.F. Immunological Response to Single Pathogen Challenge with Agents of the Bovine Respiratory Disease Complex: An RNA-Sequence Analysis of the Bronchial Lymph Node Transcriptome. PLoS ONE 2015, 10, e0131459. [Google Scholar] [CrossRef] [PubMed]
- Xiong, B.; Bayat, V.; Jaiswal, M.; Zhang, K.; Sandoval, H.; Charng, W.L.; Li, T.; David, G.; Duraine, L.; Lin, Y.Q.; et al. Crag is a GEF for rab11 required for rhodopsin trafficking and maintenance of adult photoreceptor cells. PLoS Biol. 2012, 10, 1001438. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Thery, F.; Martina, L.; Asselman, C.; Zhang, Y.; Vessely, M.; Repo, H.; Sedeyn, K.; Moschonas, G.D.; Bredow, C.; Teo, Q.W.; et al. Ring finger protein 213 assembles into a sensor for isgylated proteins with antimicrobial activity. Nat. Commun. 2021, 12, 5772. [Google Scholar] [CrossRef]
- Sugihara, M.; Morito, D.; Ainuki, S.; Hirano, Y.; Ogino, K.; Kitamura, A.; Hirata, H.; Nagata, K. The AAA+ ATPase/ubiquitin ligase mysterin stabilizes cytoplasmic lipid droplets. J. Cell Biol. 2019, 218, 949–960. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Eastman, S.W.; Chung, W.; Bieniasz, P.D. HECT ubiquitin ligases link viral and cellular PPXY motifs to the vacuolar protein-sorting pathway. J. Cell Biol. 2004, 168, 89–101. [Google Scholar] [CrossRef]
- Heidecker, G.; Lloyd, P.A.; Fox, K.; Nagashima, K.; Derse, D. Late assembly motifs of human T-cell leukemia virus type 1 and their relative roles in particle release. J. Virol. 2004, 78, 6636–6648. [Google Scholar] [CrossRef]
- Chen, H.I.; Sudol, M. The WW domain of yes-associated protein binds a proline-rich ligand that differs from the consensus established for src homology 3-binding modules. Proc. Natl. Acad. Sci. USA 1995, 92, 7819–7823. [Google Scholar] [CrossRef]
- Zhi, X.; Chen, C. WWP1: A versatile ubiquitin E3 ligase in signaling and diseases. Cell Mol. Life Sci. 2012, 69, 1425–1434. [Google Scholar] [CrossRef]
- Chen, C.S.; Bellier, A.; Kao, C.Y.; Yang, Y.L.; Chen, H.D.; Los, F.C.; Aroian, R.V. WWP-1 is a novel modulator of the DAF-2 insulin-like signaling network involved in pore-forming toxin cellular defenses in Caenorhabditis elegans. PLoS ONE 2010, 5, 9494. [Google Scholar] [CrossRef]
- Trujano-Chavez, M.Z.; Ruíz-Flores, A.; López-Ordaz, R.; Pérez-Rodríguez, P. Genetic diversity in reproductive traits of Braunvieh cattle determined with SNP markers. Vet. Med. Sci. 2022, 8, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.S.; Denicol, A.C.; Cole, J.B.; Null, D.J.; Hansen, P.J. Use of single nucleotide polymorphisms in candidate genes associated with daughter pregnancy rate for prediction of genetic merit for reproduction in Holstein Cows. Anim. Genet. 2016, 47, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Stonebraker, J.R.; Knowles, M.R.; Zariwala, M.A. A deep intronic, pathogenic variant in dnah11 causes primary ciliary dyskinesia. Am. J. Respir. Cell Mol. Biol. 2022, 67, 511–514. [Google Scholar] [CrossRef]
- Fliegauf, M.; Benzing, T.; Omran, H. When cilia go bad: Cilia defects and Ciliopathies. Nat. Rev. Mol. Cell Biol. 2007, 8, 880–893. [Google Scholar] [CrossRef]
- Dougherty, G.W.; Loges, N.T.; Klinkenbusch, J.A.; Olbrich, H.; Pennekamp, P.; Menchen, T.; Raidt, J.; Wallmeier, J.; Werner, C.; Westermann, C.; et al. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am. J. Respir. Cell Mol. Biol. 2016, 55, 213–224. [Google Scholar] [CrossRef]
- McGrath, J.; Somlo, S.; Makova, S.; Tian, X.; Brueckner, M. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell 2003, 114, 61–73. [Google Scholar] [CrossRef]
- Petronczki, M.; Lénárt, P.; Peters, J.M. Polo on the rise—From mitotic entry to cytokinesis with PLK1. Dev. Cell 2008, 14, 646–659. [Google Scholar] [CrossRef]
- Bruinsma, W.; Raaijmakers, J.A.; Medema, R.H. Switching polo-like kinase-1 on and off in time and space. Trends. Biochem. Sci. 2012, 37, 534–542. [Google Scholar] [CrossRef]
- Sumara, I.; Giménez-Abián, J.F.; Gerlich, D.; Hirota, T.; Kraft, C.; de la Torre, C.; Ellenberg, J.; Peters, J.M. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr. Biol. 2004, 14, 1712–1722. [Google Scholar] [CrossRef]
- Pan, Y.; Xue, Y.; Fei, X.; Zhao, L.; Han, L.; Su, H.; Lin, Y.; Zhou, Y.; Zhang, Y.; Xie, G.; et al. PLK1 mediates the proliferation and contraction of airway smooth muscle cells and has a role in T2-high asthma with neutrophilic inflammation model. J. Inflamm. Res. 2025, 18, 4381–4394. [Google Scholar] [CrossRef]
- Jiang, S.; Tang, D.D. PLK1 regulates mek1/2 and proliferation in airway smooth muscle cells. Respir. Res. 2015, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A.; et al. Consensus: Guidelines for diagnosis and therapy of Men Type 1 and type 2. J. Clin. Endocrinol. Metab. 2001, 86, 5658–5671. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, T.; Nagamura, Y.; Ohkura, N. MEN1 gene and its mutations: Basic and clinical implications. Cancer Sci. 2009, 100, 209–215. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, B.; Lian, A.; Zhang, X.; Wang, Y.; Zhang, Y.; Zhang, L.; Ma, J.; Gao, S.; Jin, G. Menin orchestrates macrophage reprogramming to maintain the pulmonary immune homeostasis. Cell. Rep. 2025, 44, 115219. [Google Scholar] [CrossRef]
- Trapnell, B.C.; Nakata, K.; Bonella, F.; Campo, I.; Griese, M.; Hamilton, J.; Wang, T.; Morgan, C.; Cottin, V.; McCarthy, C. Pulmonary Alveolar Proteinosis. Nat. Rev. Dis. Primers 2019, 5, 16. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, L.; Liu, Y.; Li, H.; Gong, L.; Tan, X.; Tian, J.; Pi, H.; Wang, B.; Zhao, Y.; et al. The GDF6-FTO axis modulates the innate immune and inflammatory response to human respiratory syncytial virus. iScience 2024, 27, 111038. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, H.; Xi, L.; Zhang, Y.; Zuo, X.; Yang, P.; Zhai, Z.; Wang, C. Effect of genetic variants on bleeding with rivaroxaban in pulmonary embolism patients: A multicenter prospective cohort study. Eur. Respir. J. 2024, 64 (Suppl. S68), OA5551. [Google Scholar] [CrossRef]
- National Library of Medicine. Gene: LOC617705 Deleted in Malignant Brain Tumors 1 Protein-Like Bos taurus (Domestic CATTLE). 2025. Available online: https://www.ncbi.nlm.nih.gov/gene/?term=LOC617705 (accessed on 9 April 2025).
- Keane, S.; Herring, M.; Rolny, P.; Wettergren, Y.; Ejeskär, K. Inflammation suppresses DLG2 expression decreasing inflammasome formation. J. Cancer Res. Clin. Oncol. 2022, 148, 2295–2311. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.; Ting, J.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Francoz, D.; Buczinski, S.; Apley, M. Evidence related to the use of ancillary drugs in bovine respiratory disease (anti-inflammatory and others): Are they justified or not? Vet. Clin. North Am. Food. Anim. Pract. 2012, 28, 23–38. [Google Scholar] [CrossRef]
- Grantz, J.M.; Thirumalaikumar, V.P.; Jannasch, A.H.; Andolino, C.; Taechachokevivat, N.; Avila-Granados, L.M.; Neves, R.C. The platelet and plasma proteome and targeted lipidome in postpartum dairy cows with elevated systemic inflammation. Sci. Rep. 2024, 14, 31240. [Google Scholar] [CrossRef] [PubMed]
- Drake, S.S.; Mohammadnia, A.; Zaman, A.; Gianfelice, C.; Heale, K.; Groh, A.M.R.; Hua, E.M.L.; Hintermayer, M.A.; Lu, Y.R.; Gosselin, D.; et al. Cellular rejuvenation protects neurons from inflammation-mediated cell death. Cell Rep. 2025, 44, 115298. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Yu, X.; Chen, Q.; He, X. SEMA3A inactivates the ERK/JNK signalling pathways to alleviate inflammation and oxidative stress in lipopolysaccharide-stimulated rat endothelial cells and lung tissues. Autoimmunity 2023, 56, 2200908. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Yu, J.; Jiao, Z.; Li, J.; Wu, F.; Yan, R.; Huang, X.; Chen, C. The Roles of Tissue-Resident Memory T Cells in Lung Diseases. Front. Immunol. 2021, 12, 710375. [Google Scholar] [CrossRef]
- Takamatsu, H.; Kumanogoh, A. Diverse roles for semaphorin−plexin signaling in the immune system. Trends Immunol. 2012, 33, 127–135. [Google Scholar] [CrossRef]
- McGeachie, M.J.; Wu, A.C.; Tse, S.M.; Clemmer, G.L.; Sordillo, J.; Himes, B.E.; Lasky-Su, J.; Chase, R.P.; Martinez, F.D.; Weeke, P.; et al. CTNNA3 and SEMA3D: Promising loci for asthma exacerbation identified through multiple genome-wide association studies. J. Allergy Clin. Immunol. 2015, 136, 1503–1510. [Google Scholar] [CrossRef]
- James, A.L.; Wenzel, S. Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 2007, 30, 134–155. [Google Scholar] [CrossRef]
- Gitler, A.D.; Lu, M.M.; Epstein, J.A. Plexind1 and semaphorin signaling are required in endothelial cells for cardiovascular development. Dev. Cell 2004, 7, 107–116. [Google Scholar] [CrossRef]
- Zhang, X.; Wu, Z.; Ma, K. SNCA correlates with immune infiltration and serves as a prognostic biomarker in lung adenocarcinoma. BMC Cancer 2022, 22, 406. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef]
- Celedón, J.C.; Lange, C.; Raby, B.A.; Litonjua, A.A.; Palmer, L.J.; DeMeo, D.L.; Reilly, J.J.; Kwiatkowski, D.J.; Chapman, H.A.; Laird, N.; et al. The transforming growth factor- 1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD). Hum. Mol. Genet. 2004, 13, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Denney, L.; Branchett, W.; Gregory, L.G.; Oliver, R.A.; Lloyd, C.M. Epithelial-derived TGF-β1 acts as a pro-viral factor in the lung during influenza a infection. Mucosal. Immunol. 2018, 11, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Trengove, M.C.; Ward, A.C. SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar] [PubMed] [PubMed Central]
- Kedzierski, L.; Tate, M.D.; Hsu, A.C.; Kolesnik, T.B.; Linossi, E.M.; Dagley, L.; Dong, Z.; Freeman, S.; Infusini, G.; Starkey, M.R.; et al. Suppressor of Cytokine Signaling SOCS5 ameliorates influenza infection via inhibition of EGFR signaling. eLife 2017, 6, 20444. [Google Scholar] [CrossRef]
- Sun, L.; Liu, L.; Liang, D.; Liu, L. Socs5, targeted by mir-155-5p, plays a negative regulatory role in pulmonary hypertension through inhibiting JAK2/STAT3 signaling pathway. BMC Pulm. Med. 2024, 24, 52. [Google Scholar] [CrossRef]
- Diao, X.; Zhou, J.; Wang, S.; Ma, X. Upregulation of mir-132 contributes to the pathophysiology of COPD via targeting SOCS5. Exp. Mol. Pathol. 2018, 105, 285–292. [Google Scholar] [CrossRef]
- Xue, Y.; Wu, L.; Liu, Y.; Ma, Y.; Zhang, L.; Ma, X.; Yang, Y.; Chen, J. Entpd5 induces apoptosis in lung cancer cells via regulating caspase 3 expression. PLoS ONE 2015, 10, 120046. [Google Scholar] [CrossRef]
- Schlattner, U. The complex functions of the NME family—A matter of location and molecular activity. Int. J. Mol. Sci. 2021, 22, 13083. [Google Scholar] [CrossRef]
- Šedová, L.; Buková, I.; Bažantová, P.; Petrezsélyová, S.; Prochazka, J.; Školníková, E.; Zudová, D.; Včelák, J.; Makovický, P.; Bendlová, B.; et al. Semi-lethal primary ciliary dyskinesia in rats lacking the NME7 gene. Int. J. Mol. Sci. 2021, 22, 3810. [Google Scholar] [CrossRef]
- Cho, E.H.; Huh, H.J.; Jeong, I.; Lee, N.Y.; Koh, W.; Park, H.; Ki, C. A nonsense variant in nme5 causes human primary ciliary dyskinesia with radial spoke defects. Clin. Genet. 2020, 98, 64–68. [Google Scholar] [CrossRef]
- Lee, S.; Clémentine, C.; Kim, H. Exploring the genetic factors behind the discrepancy in resistance to bovine tuberculosis between African zebu cattle and European taurine cattle. Sci. Rep. 2024, 14, 2370. [Google Scholar] [CrossRef] [PubMed]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 macrophages and their overlaps—Myth or reality? Clin. Sci. 2023, 137, 1067–1093. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhu, J.; Zhang, L.; Zhang, Z.; He, L.; Mou, Y.; Deng, Y.; Cao, Y.; Yang, P.; Su, Y.; et al. Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/transcription factor EC/IL-4 receptor α positive feedback loop in M2 macrophages. J. Allergy. Clin. Immunol. 2017, 140, 1550–1561. [Google Scholar] [CrossRef] [PubMed]
- Rehli, M.; Sulzbacher, S.; Pape, S.; Ravasi, T.; Wells, C.A.; Heinz, S.; Sollner, L.; El Chartouni, C.; Krause, S.W.; Steingrimsson, E.; et al. Transcription factor TFEC contributes to the il-4-inducible expression of a small group of genes in mouse macrophages including the granulocyte colony-stimulating factor receptor. J. Immunol. 2005, 174, 7111–7122. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, N.; Lu, Y. Fhit protein is preferentially expressed in the nucleus of monocyte-derived cells and its possible biological significance. Histol. Histopathol. 2006, 21, 915–923. [Google Scholar] [CrossRef]
- Gohari, K.; Kazemnejad, A.; Mostafaei, S.; Saberi, S.; Sheidaei, A. Chronic Obstructive Pulmonary Disease: Novel Genes Detection with Penalized Logistic Regression. Cell J. 2023, 25, 203–211. [Google Scholar] [CrossRef]
- Lin, Y.J.; Chang, J.S.; Liu, X.; Tsang, H.; Chien, W.K.; Chen, J.H.; Hsieh, H.Y.; Hsueh, K.C.; Shiao, Y.T.; Li, J.P.; et al. Genetic variants in PLCB4/PLCB1 as susceptibility loci for coronary artery aneurysm formation in Kawasaki Disease in Han Chinese in Taiwan. Sci. Rep. 2015, 5, srep14762. [Google Scholar] [CrossRef]
- Li, H.; Zhao, J.; Xing, Y.; Chen, J.; Wen, Z.; Ma, R.; Han, F.; Huang, B.; Wang, H.; Li, C.; et al. Identification of age-related characteristic genes involved in severe COVID-19 infection among elderly patients using machine learning and immune cell infiltration analysis. Biochem. Genet. 2024, 63, 1–21. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Inheritance Model 1 | p-Value 2 | BTA 3 | Mb 4 | No. Associated SNPs 5 | Positional Candidate Genes 6 |
|---|---|---|---|---|---|
| Additive | 6.37 × 10−13 | 29 | 25 | 13 | CSRP3, E2F8, IGSF22, LOC507011, MRGPRX2, PTPN5, SPTY2D1, SPTY2D1OS, TMEM86A, UEVLD, ZDHHC13 |
| Additive | 1.31 × 10−11 | 2 | 114 | 1 | - |
| Additive | 7.48 × 10−11 | 10 | 12 | 1 | DENND4A, LOC104973042 |
| Dominant | 1.09 × 10−11 | 29 | 26 | 11 | CSRP3, E2F8, IGSF22, MRGPRX2, PTPN5, SPTY2D1, SPTY2D1OS, TMEM86A, UEVLD, ZDHHC13 |
| Dominant | 2.02 × 10−10 | 10 | 12 | 1 | DENND4A, LOC104973042 |
| Dominant | 2.80 × 10−8 | 19 | 66 | 1 | LOC512869, TRNAG-CCC |
| Recessive | 7.66 × 10−14 | 2 | 28 | 1 | - |
| Recessive | 1.40 × 10−10 | 14 | 33–36 | 2 | WWP1 |
| Recessive | 7.41 × 10−9 | 4 | 114 | 6 | MACC1, LOC101907567, LOC104968411, TRNAC-GCA |
| Inheritance Model 1 | p-Value 2 | BTA 3 | Mb 4 | No. Associated SNPs 5 | Positional Candidate Genes 6 |
|---|---|---|---|---|---|
| Additive | 3.17 × 10−17 | 26 | 42 | 8 | DMBT1, FAM24A, LOC100295742, LOC100849037, LOC510536, LOC517971, LOC617705 |
| Additive | 4.60 × 10−15 | 6 | 114 | 2 | ABLIM2, MIR95 |
| Additive | 1.72 × 10−14 | 29 | 12 | 2 | DLG2 |
| Dominant | 6.79 × 10−19 | 26 | 42 | 8 | DMBT1, FAM24A, LOC100295742, LOC100849037, LOC510536, LOC617705 |
| Dominant | 1.11 × 10−14 | 29 | 12 | 2 | DLG2 |
| Dominant | 7.04 × 10−13 | 20 | 66 | 6 | LOC112443052, LOC112443058, MED10, PAPD7 |
| Recessive | 2.39 × 10−19 | 4 | 28 | 7 | MACC1 |
| Recessive | 1.02 × 10−17 | 4 | 33–36 | 17 | KIAA1324L, LOC112446533, LOC781303, SEMA3A, SEMA3D |
| Recessive | 2.01 × 10−17 | 6 | 114 | 2 | ABLIM2, MIR95 |
| Gene Set (Database) 1 | NES 2 | No. LEG 3 | Leading-Edge Genes 4 |
|---|---|---|---|
| Pre-Weaned BRD | |||
| Negative regulation of protein kinase activity (GO) | 3.92 | 19 | APOE, DUSP10, GADD45G, GSTP1, HEXIM1, HEXIM2, IGF1R, IL1B, ITGB1BP1, MEN1, NPM1, PLK1, PRKAR2A, PRKRIP1, PSEN1, SOCS2, SOCS5, STK38, TRIB2 |
| Cardiac muscle tissue development (GO) | 3.84 | 6 | CSRP3, FOXP1, MYL3, RBPJ, SMAD1, TGFB2 |
| Negative regulation of cell cycle (GO) | 3.63 | 22 | BRINP1, CDC5L, DDX39B, E2F8, FAP, HEXIM1, HEXIM2, HPGD, IL12B, LCMT1, MEN1, MLF1, MSH2, OVOL1, PKD2, PLK1, RHOB, TEX14, TGFB1, TGFB2, ZWILCH, ZWINT |
| Negative regulation of kinase activity (GO) | 3.56 | 19 | APOE, DUSP10, GADD45G, GSTP1, HEXIM1, HEXIM2, IGF1R, IL1B, ITGB1BP1, MEN1, NPM1, PLK1, PRKAR2A, PRKRIP1, PSEN1, SOCS2, SOCS5, STK38, TRIB2 |
| DNA replication (GO) | 3.54 | 33 | CDK2, DNA2, DTD1, E2F8, FHIT, GINS3, GINS4, IGF1R, KITLG, MCIDAS, MCM3, MEN1, MMS22L, NFIB, ORC2, ORC3, PDGFA, POLA2, POLD1, POLD3, POLD4, POLE2, POLG2, PRIMPOL, RBBP4, RBMS1, RFC3, RMI1, SMARCAL1, SMC3, TGFB1, TIPIN, ZPR1 |
| Post-Weaned BRD | |||
| Purine Metabolism (KEGG) | 4.43 | 39 | ADCY2, ADCY3, ADCY4, AK4, AK5, AK7, AMPD2, ENTPD1, ENTPD5, ENTPD6, ENTPD8, FHIT, GMPR, GMPR2, GUCY2C, GUCY2F, IMPDH1, NME6, NME7, PDE1C, PDE3A, PDE4B, PDE4D, PDE6A, PDE9A, POLD2, POLR1C, POLR2B, POLR2C, POLR2K, POLR3A, POLR3B, POLR3C, POLR3G, POLR3GL, POLR3K, PRIM2, RRM2, ZNRD1 |
| Cytosolic DNA sensing pathway (KEGG) | 4.17 | 16 | IL1B, IL6, NFKB1, NFKBIB, POLR1C, POLR3A, POLR3B, POLR3C, POLR3G, POLR3GL, POLR3K, RIPK1, RIPK3, TBK1, TMEM173, TREX1 |
| Organonitrogen compound metabolic process (GO) | 3.98 | 141 | AARSD1, AASS, ABAT, ABCG2, ABHD12, ABHD3, ACPP, ADAL, ADCY1, ADM, ADRB3, AK4, AK5, ALAS1, ALDH6A1, ALDH7A1, APOE, ASNS, ASNSD1, ASS1, ATP5S, ATP6V1A, BDH2, BLVRB, BTBD9, CDO1, CHIA, CHID1, CHPT1, COL4A3BP, COQ9, CPQ, CREM, CROT, CTSH, CXCL10, CXCL11, CXCL9, DARS2, DCXR, DDAH2, DDC, DEGS1, DEGS2, DHPS, DLST, DPYD, DRD1, DRD5, ECE1, EDNRA, ENOPH1, ENOSF1, ENTPD5, EXT2, FAH, FECH, FHIT, FXN, G6PC, GAL3ST1, GATA3, GATC, GBA, GGT7, GLCE, GLUD1, GMPR2, GNG7, GNS, GOT1L1, GOT2, GPLD1, GPX1, GSTA2, GSTA4, GUCY2F, HAGH, HIBCH, HPX, IL1B, IMPDH1, IP6K3, ITIH1, ITIH3, ITIH4, KCNAB2, KDSR, LYVE1, MGST2, MMAB, MSH2, MTHFD1L, MTRR, NADSYN1, NARS, NDST2, NDUFS1, NME7, NOS3, NUDT12, OAT, ODC1, OPRM1, OVGP1, PAM, PAOX, PEMT, PHGDH, PKD2, PNPO, PPARGC1A, PSEN1, PTDSS1, PTGDR, PTH, SARS, SARS2, SERINC1, SHMT2, SIRT4, SLC25A25, SMPD1, SMPDL3A, SMS, SNCA, SOD1, SPCS1, SPCS3, SPTLC1, ST6GALNAC6, STAT5B, TALDO1, TBPL1, TCN2, TGFB1, TH, TYMS, UMPS, WARS, XDH |
| Cell cycle (R) | 3.96 | 109 | ALMS1, ANAPC1, ANAPC4, ATR, ATRIP, B9D2, BRCA1, BUB1, BUB3, CCND2, CDC14A, CDC25C, CDC45, CDK4, CENPA, CENPN, CENPO, CEP192, CHEK2, CLASP1, CUL1, DIDO1, DYNC1H1, DYRK1A, E2F2, E2F3, GINS1, GINS2, HAUS2, HIST1H2BA, HIST1H2BB, HIST1H2BI, HIST1H2BJ, HIST1H2BL, HIST1H2BN, HIST2H2AA4, HIST2H2AC, HIST2H2BE, KIF18A, KIF23, LIN52, LIN54, MAD1L1, MCM3, MCM7, MIS18A, MIS18BP1, MYC, NDEL1, NEDD1, OFD1, ORC3, ORC6, PCNT, POLD2, POT1, PPP2R5A, PPP2R5C, PPP2R5D, PPP2R5E, PRIM2, PRKAR2B, PSMA5, PSMA6, PSMB10, PSMB5, PSMB8, PSMB9, PSMC3, PSMC4, PSMC5, PSMC6, PSMD12, PSMD4, PSMD7, PSMD8, PSME1, PSME2, PSME4, PTTG1, RANGAP1, RB1, REC8, RFC2, RFC3, RPA1, RPA3, RRM2, SEH1L, SSNA1, STAG1, STAG2, STAG3, SYCP3, SYNE1, SYNE2, TERF1, TERF2, TFDP1, TINF2, TUBB, TUBG1, TUBGCP6, TYMS, UBE2I, WEE1, XPO1, ZW10, ZWILCH |
| Purine ribonucleoside metabolic process (GO) | 3.95 | 18 | ADAL, AK4, AK5, ATP5S, ATP6V1A, COQ9, CROT, ENTPD5, FXN, GMPR2, IMPDH1, IP6K3, NME7, PPARGC1A, PTGDR, SLC25A25, SNCA, TGFB1 |
| Age and Analysis Where Genes Were Shared 1 | No. Genes 2 | Positional Candidate or Leading-Edge Genes 3 |
|---|---|---|
| PCG & LEG in Pre-Weaned Heifers | 1 | SOCS5 |
| PCG & LEG in Post-Weaned Heifers | 11 | ALMS1, C6, ENTPD5, FHIT, LOC534742, NME7, SEMA3C, STAG1, SYNDIG1L, TFEC, XPO1 |
| PCG in Pre- and Post-Weaned Heifers | 16 | GPNMB, IGF2BP3, LOC100138586, LOC101907567, LOC101907914, LOC104968411, LOC107132385, LOC107132393, MACC1, MALSU1, NUPL2, RAPGEF5, TENM3, TFEC, TRNAC-GCA, TRNAK-UUU |
| LEG in Pre- and Post-Weaned Heifers | 36 | APOE, ARL3, ARRB1, C4A, CFB, CFH, CSRP3, DCPS, DDX39B, DUSP10, E2F8, FHIT, FOXP1, IL12B, IL1B, ISG15, LTA, LTF, MCM3, MSH2, NOD2, ORC3, PKD2, PSEN1, RBM22, RBM4, RBPJ, RFC3, RHOA, SMARCAL1, SOCS2, TGFB1, TGFB2, TNF, USP4, ZWILCH |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrick, A.L.; Kiser, J.N.; White, S.N.; Neibergs, H.L. Genomic Regions Associated with Respiratory Disease in Holstein Calves in the Southern United States. Genes 2025, 16, 741. https://doi.org/10.3390/genes16070741
Herrick AL, Kiser JN, White SN, Neibergs HL. Genomic Regions Associated with Respiratory Disease in Holstein Calves in the Southern United States. Genes. 2025; 16(7):741. https://doi.org/10.3390/genes16070741
Chicago/Turabian StyleHerrick, Allison L., Jennifer N. Kiser, Stephen N. White, and Holly L. Neibergs. 2025. "Genomic Regions Associated with Respiratory Disease in Holstein Calves in the Southern United States" Genes 16, no. 7: 741. https://doi.org/10.3390/genes16070741
APA StyleHerrick, A. L., Kiser, J. N., White, S. N., & Neibergs, H. L. (2025). Genomic Regions Associated with Respiratory Disease in Holstein Calves in the Southern United States. Genes, 16(7), 741. https://doi.org/10.3390/genes16070741