
Optical Genome Mapping: A New Tool for Cytogenomic Analysis


Abstract
1. Introduction
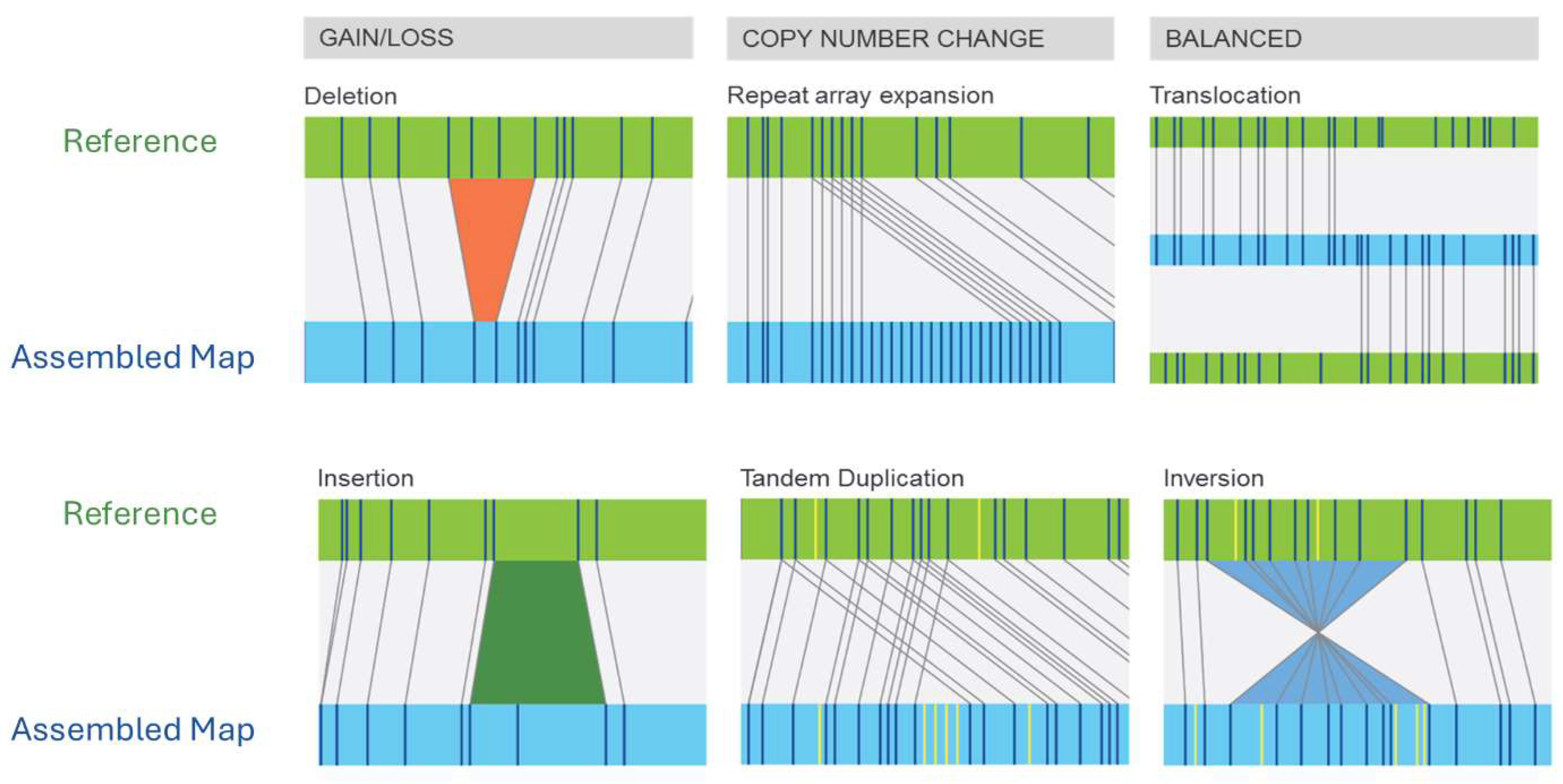
2. Technological Foundations of OGM
Principles and Methodology
3. Clinical Applications of OGM
3.1. Hematologic Malignancies and Solid Tumors
3.2. Constitutional Studies
4. Advantages of OGM
4.1. Resolution of Complex Genomic Rearrangements
4.2. Analysis of Repetitive Regions and Tandem Repeats
4.3. Time and Cost-Effectiveness in Clinical Settings
5. Limitations of OGM
5.1. Technical Limitations and Areas for Improvement
5.2. Data Interpretation and Variant Classification
5.3. Integration into Existing Clinical Workflows
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boveri, T. Zur Frage der Entstehung Maligner Tumoren (On the Problem of the Origin of Malignant Tumors); Gustav Fischer: Jena, Germany, 1914. [Google Scholar]
- Lejeune, J.; Gautier, M.; Turpin, R. Etude des chromosomes somatiques de neuf enfants mongoliens. Compt. Rend. 1959, 248, 1721–1722. [Google Scholar]
- Hirschhorn, K.; Kolodny, R.; Hashem, N.; Bach, F. Mitogenic action of phytohaemagglutinin. Lancet 1963, 282, 305–306. [Google Scholar] [CrossRef]
- Moorhead, P.S.; Nowell, P.C.; Mellman, W.J.; Battips, D.M.; Hungerford, D.A. Chromosome preparations of leukocytes cultured from human peripheral blood. Exp. Cell Res. 1960, 20, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.C. Phytohemagglutinin: An initiator of mitosis in cultures of normal human leukocytes. Cancer Res. 1960, 20, 462–466. [Google Scholar] [PubMed]
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109. [Google Scholar] [PubMed]
- Pearmain, G.; Lycette, R.R.; Fitzgerald, P.H. Tuberculin-induced mitosis in peripheral blood leucocytes. Lancet 1963, 283, 637–638. [Google Scholar] [CrossRef]
- Caspersson, T.; Zech, L.; Johansson, C. Differential binding of alkylating fluorochromes in human chromosomes. Exp. Cell Res. 1970, 60, 315–319. [Google Scholar] [CrossRef]
- Drets, M.E.; Shaw, M.W. Specific banding patterns of human chromosomes. Proc. Natl. Acad. Sci. USA 1971, 68, 2073–2077. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.R.; Merrick, S.; Lubs, H.A. Identification of Each Human Chromosome with a Modified Giemsa Stain. Science 1971, 173, 821–822. [Google Scholar] [CrossRef]
- Seabright, M. A rapid banding technique for human chromosomes. Lancet 1971, 2, 971–972. [Google Scholar] [CrossRef]
- Sumner, A.T.; Evans, H.J.; Buckland, R.A. New technique for distinguishing between human chromosomes. Nat. New Biol. 1971, 232, 31–32. [Google Scholar] [CrossRef]
- Hirschhorn, K.; Lucas, M.; Wallace, I. Precise identification of various chromosomal abnormalities. Ann. Hum. Genet. 1973, 36, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Kallioniemi, A.; Kallioniemi, O.P.; Sudar, D.; Rutovitz, D.; Gray, J.W.; Waldman, F.; Pinkel, D. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.; Dunn, T.M.; Kaffe, S.; Kardon, N.; Hirschhorn, K. Clinical applications of comparative genomic hybridization. Genet. Med. 1998, 1, 4–12. [Google Scholar] [CrossRef]
- Pinkel, D.; Segraves, R.; Sudar, D.; Clark, S.; Poole, I.; Kowbel, D.; Collins, C.; Kuo, W.L.; Chen, C.; Zhai, Y.; et al. High resolution analysis of DNA copy number variation using comparative genomic hybridization to microarrays. Nat. Genet. 1998, 20, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Solinas-Toldo, S.; Lampel, S.; Stilgenbauer, S.; Nickolenko, J.; Benner, A.; Dohner, H.; Cremer, T.; Lichter, P. Matrix-based comparative genomic hybridization: Biochips to screen for genomic imbalances. Genes Chromosomes Cancer 1997, 20, 399–407. [Google Scholar] [CrossRef]
- Manning, M.; Hudgins, L.; Professional, P.; Guidelines, C. Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities. Genet. Med. 2010, 12, 742–745. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Levy, B.; Wapner, R. Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 2018, 109, 201–212. [Google Scholar] [CrossRef]
- Li, M.M.; Monzon, F.A.; Biegel, J.A.; Jobanputra, V.; Laffin, J.J.; Levy, B.; Leon, A.; Miron, P.; Rossi, M.R.; Toruner, G.; et al. A multicenter, cross-platform clinical validation study of cancer cytogenomic arrays. Cancer Genet. 2015, 208, 525–536. [Google Scholar] [CrossRef]
- Iqbal, M.A.; Broeckel, U.; Levy, B.; Skinner, S.; Sahajpal, N.S.; Rodriguez, V.; Stence, A.; Awayda, K.; Scharer, G.; Skinner, C.; et al. Multisite Assessment of Optical Genome Mapping for Analysis of Structural Variants in Constitutional Postnatal Cases. J. Mol. Diagn. 2023, 25, 175–188. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Puiggros, A.; Granada, I.; Raca, G.; Rack, K.; Mallo, M.; Dewaele, B.; Smith, A.C.; Akkari, Y.; Levy, B.; et al. Integration of Optical Genome Mapping in the Cytogenomic and Molecular Work-Up of Hematological Malignancies: Expert Recommendations From the International Consortium for Optical Genome Mapping. Am. J. Hematol. 2025, 100, 1029–1048. [Google Scholar] [CrossRef]
- Levy, B.; Baughn, L.B.; Akkari, Y.; Chartrand, S.; LaBarge, B.; Claxton, D.; Lennon, P.A.; Cujar, C.; Kolhe, R.; Kroeger, K.; et al. Optical genome mapping in acute myeloid leukemia: A multicenter evaluation. Blood. Adv. 2023, 7, 1297–1307. [Google Scholar] [CrossRef]
- Levy, B.; Kanagal-Shamanna, R.; Sahajpal, N.S.; Neveling, K.; Rack, K.; Dewaele, B.; Olde Weghuis, D.; Stevens-Kroef, M.; Puiggros, A.; Mallo, M.; et al. A framework for the clinical implementation of optical genome mapping in hematologic malignancies. Am. J. Hematol. 2024, 99, 642–661. [Google Scholar] [CrossRef]
- Levy, B.; Liu, J.; Iqbal, M.A.; DuPont, B.; Sahajpal, N.; Ho, M.; Yu, J.; Brody, S.J.; Ganapathi, M.; Rajkovic, A.; et al. Multisite Evaluation and Validation of Optical Genome Mapping for Prenatal Genetic Testing. J. Mol. Diagn. 2024, 26, 906–916. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Barseghyan, H.; Kolhe, R.; Hastie, A.; Chaubey, A. Optical Genome Mapping as a Next-Generation Cytogenomic Tool for Detection of Structural and Copy Number Variations for Prenatal Genomic Analyses. Genes 2021, 12, 398. [Google Scholar] [CrossRef] [PubMed]
- Toruner, G.A.; Hu, S.; Loghavi, S.; OK, C.Y.; Tang, Z.; Wei, Q.; Kanagal-Shamanna, R.; Medeiros, L.J.; Tang, G. Clinical Utility of Optical Genome Mapping as an Additional Tool in a Standard Cytogenetic Workup in Hematological Malignancies. Cancers 2025, 17, 1436. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, D.C.; Li, X.; Hernandez, L.I.; Ramnarain, S.P.; Huff, E.J.; Wang, Y.K. Ordered restriction maps of Saccharomyces cerevisiae chromosomes constructed by optical mapping. Science 1993, 262, 110–114. [Google Scholar] [CrossRef]
- Kidd, J.M.; Cooper, G.M.; Donahue, W.F.; Hayden, H.S.; Sampas, N.; Graves, T.; Hansen, N.; Teague, B.; Alkan, C.; Antonacci, F.; et al. Mapping and sequencing of structural variation from eight human genomes. Nature 2008, 453, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Nogin, Y.; Bar-Lev, D.; Hanania, D.; Detinis Zur, T.; Ebenstein, Y.; Yaakobi, E.; Weinberger, N.; Shechtman, Y. Design of optimal labeling patterns for optical genome mapping via information theory. Bioinformatics 2023, 39, btad601. [Google Scholar] [CrossRef]
- Akkari, Y.M.N.; Baughn, L.B.; Dubuc, A.M.; Smith, A.C.; Mallo, M.; Dal Cin, P.; Diez Campelo, M.; Gallego, M.S.; Granada Font, I.; Haase, D.T.; et al. Guiding the global evolution of cytogenetic testing for hematologic malignancies. Blood 2022, 139, 2273–2284. [Google Scholar] [CrossRef]
- Akkari, Y.; Dobin, S.; Best, R.G.; Leung, M.L. Exploring Current Challenges in the Technologist Workforce of Clinical Genomics Laboratories in the United States. Genet. Med. 2023, 1, 100806. [Google Scholar] [CrossRef]
- Fang, H.; Eacker, S.M.; Wu, Y.; Neufeld-Kaiser, W.; Laurino, M.; Keel, S.; Horwitz, M.S.; Liu, Y.J. Genetic and functional characterization of inherited complex chromosomal rearrangements in a family with multisystem anomalies. Genet. Med. Open. 2025, 3, 103423. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, S.A.; Gagnon, V.; Béliveau, F.; Lavallée, S.; Collin, V.; Hébert, J. Unveiling the Complexity of KMT2A Rearrangements in Acute Myeloid Leukemias with Optical Genome Mapping. Cancers 2024, 16, 4171. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Toruner, G.A.; Thakral, B.; Patel, K.P.; Pemmaraju, N.; Wang, S.A.; Kanagal-Shamanna, R.; Tang, G.; Issa, G.C.; Loghavi, S.; et al. Cryptic KMT2A::AFDN Fusion Due to AFDN Insertion into KMT2A in a Patient with Acute Monoblastic Leukemia. Genes 2025, 16, 317. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.; Li, M.; Zhang, H.; Chang, J.; Qi, Q.; Zhou, X.; Guo, J.; Wang, Y.; Mao, X.; Hao, N.; et al. Optical genome mapping to decipher the chromosomal aberrations in families seeking for preconception genetic counseling. Sci. Rep. 2025, 15, 2614. [Google Scholar] [CrossRef]
- Yang, H.; Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Tang, Z.; Wei, Y.; Kadia, T.; Chien, K.; Rush, D.; Nguyen, H.; et al. High-resolution structural variant profiling of myelodysplastic syndromes by optical genome mapping uncovers cryptic aberrations of prognostic and therapeutic significance. Leukemia 2022, 36, 2306–2316. [Google Scholar] [CrossRef]
- Singh, H.; Sahajpal, N.S.; Mondal, A.K.; Burke, S.L.; Farmaha, J.; Alptekin, A.; Vashisht, A.; Jones, K.; Vashisht, V.; Kolhe, R. Clinical Utility of Optical Genome Mapping for Improved Cytogenomic Analysis of Gliomas. Biomedicines 2024, 12, 1659. [Google Scholar] [CrossRef]
- Barford, R.G.; Whittle, E.; Weir, L.; Fong, F.C.; Goodman, A.; Hartley, H.E.; Allinson, L.M.; Tweddle, D.A. Use of Optical Genome Mapping to Detect Structural Variants in Neuroblastoma. Cancers 2023, 15, 5233. [Google Scholar] [CrossRef]
- Paulraj, P.; Barrie, E.; Jackson-Cook, C. Optical genome mapping reveals balanced and unbalanced genetic changes associated with tumor-forming potential in an early-stage prostate cancer epithelial subline (M2205). Mol. Genet. Genom. Med. 2023, 12, e2307. [Google Scholar] [CrossRef]
- Goldrich, D.Y.; LaBarge, B.; Chartrand, S.; Zhang, L.; Sadowski, H.B.; Zhang, Y.; Pham, K.; Way, H.; Lai, C.-Y.J.; Pang, A.W.C.; et al. Identification of Somatic Structural Variants in Solid Tumors by Optical Genome Mapping. J. Pers. Med. 2021, 11, 142. [Google Scholar] [CrossRef]
- Sahajpal, N.S.; Mondal, A.K.; Hastie, A.; Chaubey, A.; Kolhe, R. Optical Genome Mapping for Oncology Applications. Curr. Protoc. 2023, 3, e910. [Google Scholar] [CrossRef]
- Broeckel, U.; Iqbal, M.A.; Levy, B.; Sahajpal, N.; Nagy, P.L.; Scharer, G.; Rodriguez, V.; Bossler, A.; Stence, A.; Skinner, C.; et al. Detection of Constitutional Structural Variants by Optical Genome Mapping: A Multisite Study of Postnatal Samples. J. Mol. Diagn. 2024, 26, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Mantere, T.; Neveling, K.; Pebrel-Richard, C.; Benoist, M.; van der Zande, G.; Kater-Baats, E.; Baatout, I.; van Beek, R.; Yammine, T.; Oorsprong, M.; et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am. J. Hum. Genet. 2021, 108, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Guo, J.; Sang, H.; Yan, J.; Chang, H.; Liu, T.; Dong, H.; Kong, M.; Tian, Y.; Jiang, L. Case Report: Optical genome mapping enables identification of complex balanced chromosomal rearrangements. Front. Genet. 2025, 16, 1555485. [Google Scholar] [CrossRef] [PubMed]
- Orlando, V.; Di Tommaso, S.; Alesi, V.; Loddo, S.; Genovese, S.; Catino, G.; Martucci, L.; Roberti, M.C.; Trivisano, M.; Dentici, M.L.; et al. A Complex Genomic Rearrangement Resulting in Loss of Function of SCN1A and SCN2A in a Patient with Severe Developmental and Epileptic Encephalopathy. Int. J. Mol. Sci. 2022, 23, 12900. [Google Scholar] [CrossRef]
- Xie, M.; Xue, J.; Zhang, Y.; Zhou, Y.; Yu, Q.; Li, H.; Li, Q. Combination of trio-based whole exome sequencing and optical genome mapping reveals a cryptic balanced translocation that causes unbalanced chromosomal rearrangements in a family with multiple anomalies. Front. Genet. 2023, 14, 1248544. [Google Scholar] [CrossRef]
- Zhang, S.; Pei, Z.; Lei, C.; Zhu, S.; Deng, K.; Zhou, J.; Yang, J.; Lu, D.; Sun, X.; Xu, C.; et al. Detection of cryptic balanced chromosomal rearrangements using high-resolution optical genome mapping. J. Med. Genet. 2023, 60, 274–284. [Google Scholar] [CrossRef]
- Cuillerier, A.; Del Gobbo, G.F.; Mackay, L.; Wall, E.; Couse, M.; McDonell, L.M.; Cloutier, M.; Danzi, M.C.; Warman-Chardon, J.; Bourque, P.R.; et al. FGF14 GAA Intronic Expansion in Unsolved Adult-Onset Ataxia in the Care4Rare Canada Consortium. Ann. Clin. Transl. Neurol. 2025, 12, 1118–1125. [Google Scholar] [CrossRef]
- Mutlu, M.B.; Karakaya, T.; Celebi, H.B.G.; Duymus, F.; Seyhan, S.; Yilmaz, S.; Yis, U.; Atik, T.; Yetkin, M.F.; Gumus, H. Utility of Optical Genome Mapping in Repeat Disorders. Clin. Genet. 2025, 107, 188–195. [Google Scholar] [CrossRef]
- van der Sanden, B.; Neveling, K.; Shukor, S.; Gallagher, M.D.; Lee, J.; Burke, S.L.; Pennings, M.; van Beek, R.; Oorsprong, M.; Kater-Baats, E.; et al. Optical genome mapping enables accurate testing of large repeat expansions. Genome. Res. 2025, 35, 810–823. [Google Scholar] [CrossRef]
- Zarouchlioti, C.; Efthymiou, S.; Facchini, S.; Dominik, N.; Bhattacharyya, N.; Liu, S.; Costa, M.A.; Szabo, A.; Sadan, A.N.; Jun, A.S.; et al. Tissue-specific TCF4 triplet repeat instability revealed by optical genome mapping. EBioMedicine 2024, 108, 105328. [Google Scholar] [CrossRef]
- Xie, M.; Zheng, Z.J.; Zhou, Y.; Zhang, Y.X.; Li, Q.; Tian, L.Y.; Cao, J.; Xu, Y.T.; Ren, J.; Yu, Q.; et al. Prospective Investigation of Optical Genome Mapping for Prenatal Genetic Diagnosis. Clin. Chem. 2024, 70, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Sahajpal, N.S.; Mondal, A.K.; Fee, T.; Hilton, B.; Layman, L.; Hastie, A.R.; Chaubey, A.; DuPont, B.R.; Kolhe, R. Clinical Validation and Diagnostic Utility of Optical Genome Mapping in Prenatal Diagnostic Testing. J. Mol. Diagn. 2023, 25, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, H.; Zhang, Q.; Tan, J.; Zhou, R.; Ji, X.; Luo, C.; Meng, L.; Liu, A.; Wang, Y.; et al. Optical Genome Mapping for Prenatal Diagnosis in Fetuses With Structural Anomalies. Prenat. Diagn. 2025, 45, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Rao, H.; Zhang, H.; Zou, Y.; Ma, P.; Huang, T.; Yuan, H.; Zhou, J.; Lu, W.; Li, Q.; Huang, S.; et al. Analysis of chromosomal structural variations in patients with recurrent spontaneous abortion using optical genome mapping. Front. Genet. 2023, 14, 1248755. [Google Scholar] [CrossRef]
- Ghabrial, J.; Stinnett, V.; Ribeiro, E.; Klausner, M.; Morsberger, L.; Long, P.; Middlezong, W.; Xian, R.; Gocke, C.; Lin, M.T.; et al. Diagnostic and Prognostic/Therapeutic Significance of Comprehensive Analysis of Bone and Soft Tissue Tumors Using Optical Genome Mapping and Next-Generation Sequencing. Mod. Pathol. 2025, 38, 100684. [Google Scholar] [CrossRef]
- Lestringant, V.; Guermouche-Flament, H.; Jimenez-Pocquet, M.; Gaillard, J.B.; Penther, D. Cytogenetics in the management of hematological malignancies: An overview of alternative technologies for cytogenetic characterization. Curr. Res. Transl. Med. 2024, 72, 103440. [Google Scholar] [CrossRef]
- Loghavi, S.; Wei, Q.; Ravandi, F.; Quesada, A.E.; Routbort, M.J.; Hu, S.; Toruner, G.A.; Wang, S.A.; Wang, W.; Miranda, R.N.; et al. Optical genome mapping improves the accuracy of classification, risk stratification, and personalized treatment strategies for patients with acute myeloid leukemia. Am. J. Hematol. 2024, 99, 1959–1968. [Google Scholar] [CrossRef]
- Neveling, K.; Mantere, T.; Vermeulen, S.; Oorsprong, M.; van Beek, R.; Kater-Baats, E.; Pauper, M.; van der Zande, G.; Smeets, D.; Weghuis, D.O.; et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 2021, 108, 1423–1435. [Google Scholar] [CrossRef]
- Valkama, A.; Vorimo, S.; Tervasmaki, A.; Rasanen, H.; Savolainen, E.R.; Pylkas, K.; Mantere, T. Structural Variant Analysis of Complex Karyotype Myelodysplastic Neoplasia Through Optical Genome Mapping. Genes Chromosomes Cancer. 2025, 64, e70024. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Q.; Wang, Y.; Zhou, R.; Ji, X.; Meng, L.; Luo, C.; Liu, A.; Jiao, J.; Chen, H.; et al. Optical Genome Mapping for Chromosomal Aberrations Detection—False-Negative Results and Contributing Factors. Diagnostics 2024, 14, 165. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, S.; Lemmers, R.J.L.F.; Vishnu, V.Y.; Dominik, N.; Perrone, B.; Facchini, S.; Vegezzi, E.; Ravaglia, S.; Wilson, L.; van der Vliet, P.J.; et al. Optical Genome Mapping for the Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy: Advancement and Challenges. Biomolecules 2023, 13, 1567. [Google Scholar] [CrossRef] [PubMed]
- Guruju, N.M.; Jump, V.; Lemmers, R.; Van Der Maarel, S.; Liu, R.; Nallamilli, B.R.; Shenoy, S.; Chaubey, A.; Koppikar, P.; Rose, R.; et al. Molecular Diagnosis of Facioscapulohumeral Muscular Dystrophy in Patients Clinically Suspected of FSHD Using Optical Genome Mapping. Neurol. Genet. 2023, 9, e200107. [Google Scholar] [CrossRef] [PubMed]
- Kovanda, A.; Lovrečić, L.; Rudolf, G.; Babic Bozovic, I.; Jaklič, H.; Leonardis, L.; Peterlin, B. Evaluation of Optical Genome Mapping in Clinical Genetic Testing of Facioscapulohumeral Muscular Dystrophy. Genes 2023, 14, 2166. [Google Scholar] [CrossRef]
- Vishnu, V.Y.; Lemmers, R.; Reyaz, A.; Mishra, R.; Ahmad, T.; van der Vliet, P.J.; Kretkiewicz, M.M.; Macken, W.L.; Efthymiou, S.; Dominik, N.; et al. The first genetically confirmed cohort of Facioscapulohumeral Muscular Dystrophy from Northern India. Eur. J. Hum. Genet. 2024, 32, 1053–1064. [Google Scholar] [CrossRef]
- Facchini, S.; Dominik, N.; Manini, A.; Efthymiou, S.; Currò, R.; Rugginini, B.; Vegezzi, E.; Quartesan, I.; Perrone, B.; Kutty, S.K.; et al. Optical Genome Mapping Enables Detection and Accurate Sizing of RFC1 Repeat Expansions. Biomolecules 2023, 13, 1546. [Google Scholar] [CrossRef]
- Ciobanu, C.-G.; Nucă, I.; Popescu, R.; Antoci, L.-M.; Caba, L.; Ivanov, A.V.; Cojocaru, K.-A.; Rusu, C.; Mihai, C.-T.; Pânzaru, M.-C. Narrative Review: Update on the Molecular Diagnosis of Fragile X Syndrome. Int. J. Mol. Sci. 2023, 24, 9206. [Google Scholar] [CrossRef]
- Straits Research. Optical Genome Mapping Market Size, Share & Trends Analysis Report By Product & Services (Instruments, Consumables and Reagents, Software, Services), by Application (Structural Variant Detection, Genome Assembly, Microbial Strain Typing, Others), by End-User (Biotechnology and Pharmaceutical Companies, Clinical Laboratories, Academic Research Institutes, Others) and by Region (North America, Europe, APAC, Middle East and Africa, LATAM) Forecasts, 2024–2032. 2024. Available online: https://straitsresearch.com/report/optical-genome-mapping-market#:~:text=China%20optical%20genome%20mapping%20market,market%20growth%20in%20the%20country (accessed on 22 June 2025).
- Nguyen-Khac, F.; Bidet, A.; Chapiro, E.; Lefebvre, C.; Michaux, L.; Troadec, M.B. Cytogenetics in the management of hematological malignancies: Guidelines from the Groupe Francophone de Cytogenetique Hematologique. Curr. Res. Transl. Med. 2023, 71, 103411. [Google Scholar] [CrossRef]
- Al-Amer, O.M.; Khubrani, Y. Cytogenetics and the Revolution of Optical Genome Mapping in the Diagnosis of Diseases. Discov. Med. 2024, 36, 1780–1788. [Google Scholar] [CrossRef]
- Smith, A.C.; Hoischen, A.; Raca, G. Cytogenetics Is a Science, Not a Technique! Why Optical Genome Mapping Is So Important to Clinical Genetic Laboratories. Cancers 2023, 15, 5470. [Google Scholar] [CrossRef]
- Barseghyan, H.; Pang, A.W.C.; Clifford, B.; Serrano, M.A.; Chaubey, A.; Hastie, A.R. Comparative Benchmarking of Optical Genome Mapping and Chromosomal Microarray Reveals High Technological Concordance in CNV Identification and Additional Structural Variant Refinement. Genes 2023, 14, 1868. [Google Scholar] [CrossRef] [PubMed]
- Papenhausen, P.; Schwartz, S.; Risheg, H.; Keitges, E.; Gadi, I.; Burnside, R.D.; Jaswaney, V.; Pappas, J.; Pasion, R.; Friedman, K.; et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am. J. Med. Genet. A 2011, 155, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.T.; Hastie, A.; Lin, C.; Ehrlich, D.; Das, S.K.; Austin, M.D.; Deshpande, P.; Cao, H.; Nagarajan, N.; Xiao, M.; et al. Genome mapping on nanochannel arrays for structural variation analysis and sequence assembly. Nat. Biotechnol. 2012, 30, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.C.; Lai, Y.Y.; Lam, E.T.; Kwok, T.P.; Leung, A.K.; Poon, A.; Mostovoy, Y.; Hastie, A.R.; Stedman, W.; Anantharaman, T.; et al. Genome-Wide Structural Variation Detection by Genome Mapping on Nanochannel Arrays. Genetics 2016, 202, 351–362. [Google Scholar] [CrossRef]
- ISCN 2024—An International System for Human Cytogenomic Nomenclature (2024). Cytogenet. Genome. Res. 2024, 164, 1–224. [CrossRef]
- Tuefferd, M.; De Bondt, A.; Van Den Wyngaert, I.; Talloen, W.; Verbeke, T.; Carvalho, B.; Clevert, D.A.; Alifano, M.; Raghavan, N.; Amaratunga, D.; et al. Genome-wide copy number alterations detection in fresh frozen and matched FFPE samples using SNP 6.0 arrays. Genes Chromosomes Cancer 2008, 47, 957–964. [Google Scholar] [CrossRef]

{kind=link}
| Application | Germline DNA Analysis | Somatic DNA Analysis |
|---|---|---|
| Data collected | 400 Gbp | 1.5 Tbp |
| Coverage setting | 100× | 400× |
| Effective coverage | 80× | 300× |
| Variant allele frequency | ≥50% | ≥5% |
| Analysis pipeline | De Novo Assembly | |
| Resolution by variant type at >90% sensitivity | ||
| Insertions | >500 bp | >5 kbp |
| Deletions | >700 bp | >7 kbp |
| Repeat expansion/contractions | >500 bp | >5 kbp |
| Duplications | >30 kbp | >150 kbp |
| Translocations | >70 kbp | >70 kbp |
| Inversions | >30 kbp | >70 kbp |
| G-Banded Chromosome Analysis | FISH | CMA | GS/ES | OGM | |
|---|---|---|---|---|---|
| Whole genome coverage | √ | X | √ | +/− | √ |
| AOH | X | X | √ | +/− | √ |
| Repeat Expansions/Contractions | X | X | X | +/− | √ |
| Resolution | 5–10 Mb | 60 Kb | 25 Kb | SNV | 500 bp |
| Limit of Detection | Single Cell | Single Cell | ~10% | ~1–5% | ~20% |
| Detects Balanced SVs | +/− | +/− | X | √ | √ |
| Bioinformatics required | No | No | No | Yes | Yes |
| Turnaround Time | 5–28 days | 24 h–5 days | ~7 days | ~4 weeks | ~7 days |
| Cost | $ | $ | $$ | $$$$ | $$ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levy, B.; Burnside, R.D.; Akkari, Y. Optical Genome Mapping: A New Tool for Cytogenomic Analysis. Genes 2025, 16, 924. https://doi.org/10.3390/genes16080924
Levy B, Burnside RD, Akkari Y. Optical Genome Mapping: A New Tool for Cytogenomic Analysis. Genes. 2025; 16(8):924. https://doi.org/10.3390/genes16080924
Chicago/Turabian StyleLevy, Brynn, Rachel D. Burnside, and Yassmine Akkari. 2025. "Optical Genome Mapping: A New Tool for Cytogenomic Analysis" Genes 16, no. 8: 924. https://doi.org/10.3390/genes16080924
APA StyleLevy, B., Burnside, R. D., & Akkari, Y. (2025). Optical Genome Mapping: A New Tool for Cytogenomic Analysis. Genes, 16(8), 924. https://doi.org/10.3390/genes16080924