





Int. J. Mol. Sci. 2022, 23(13), 7370; https://doi.org/10.3390/ijms23137370 - 1 Jul 2022
Cited by 5 | Viewed by 3149
Abstract
►
Show Figures
Wheat leaf rust (caused by Puccinia triticina Erikss.) is among the major diseases of common wheat. The lack of resistance genes to leaf rust has limited the development of wheat cultivars. Wheat–Agropyron cristatum (A. cristatum) 2P addition line II-9-3 has
[...] Read more.
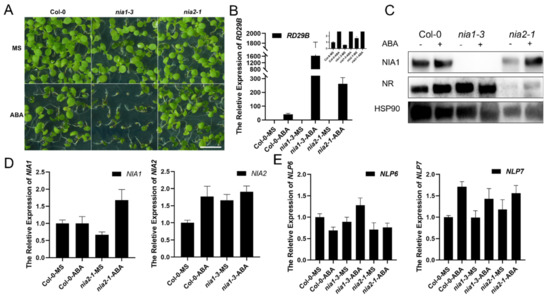
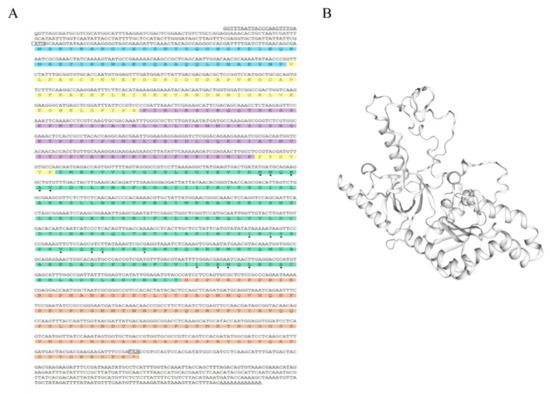
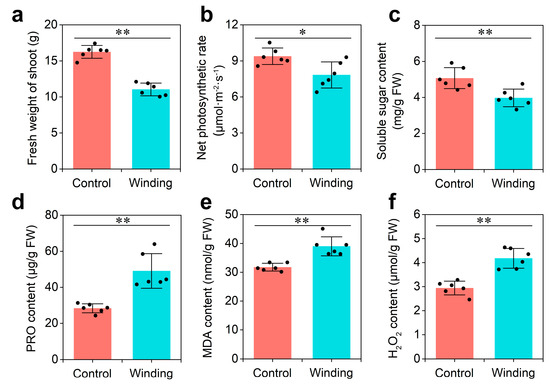
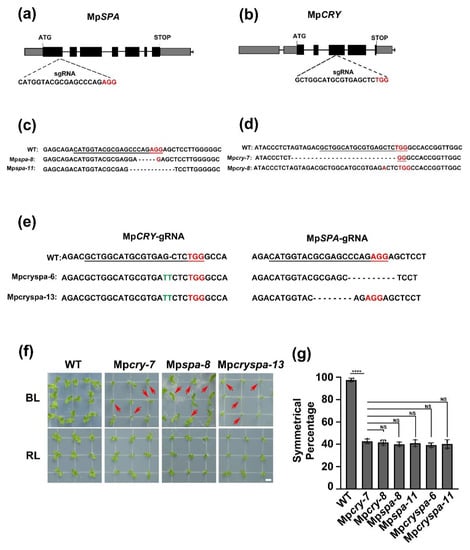
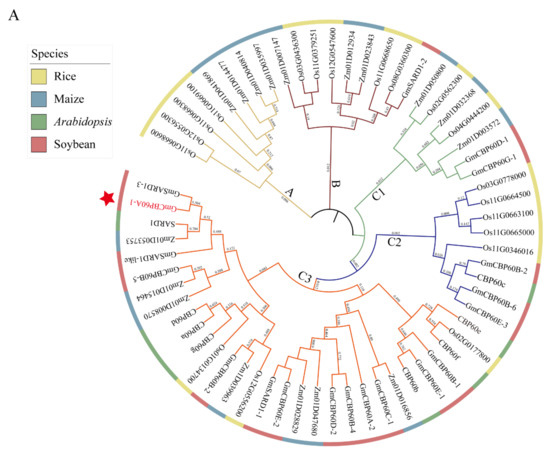
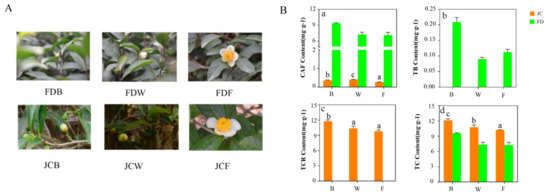
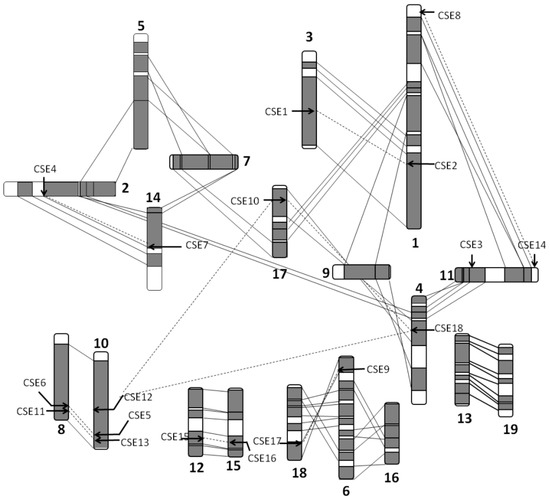
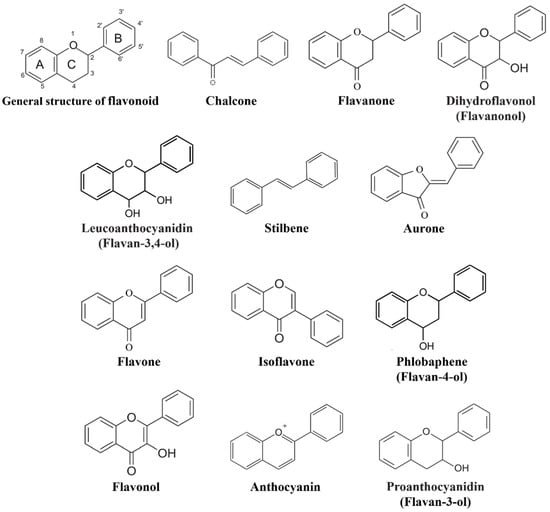
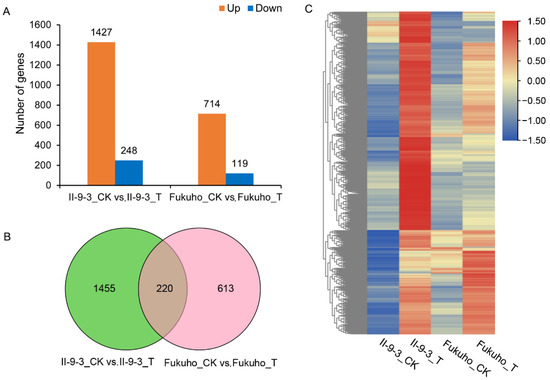
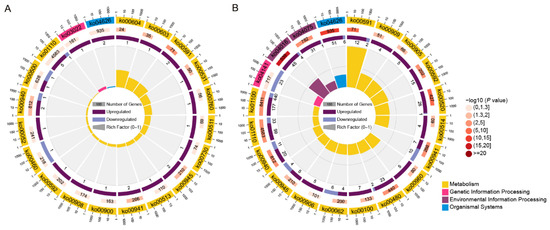
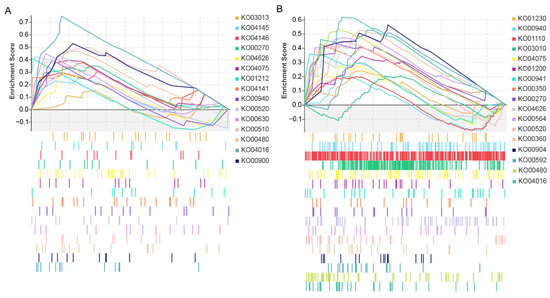
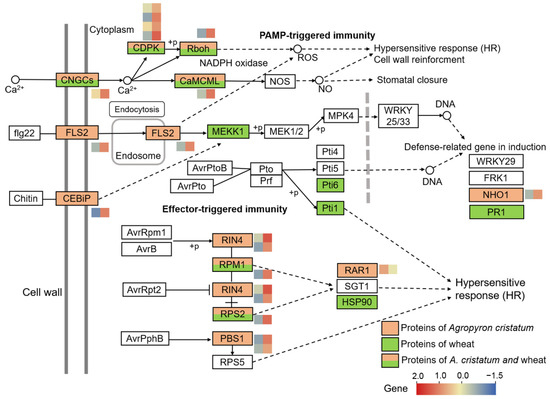
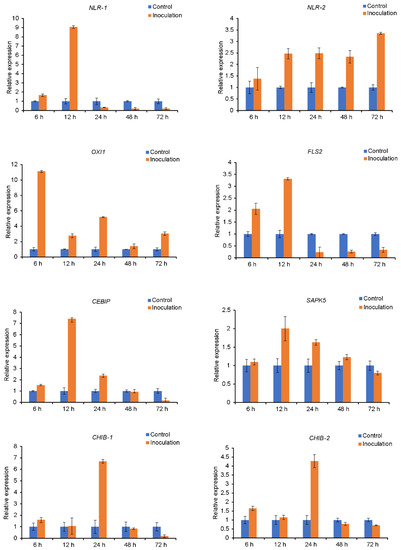
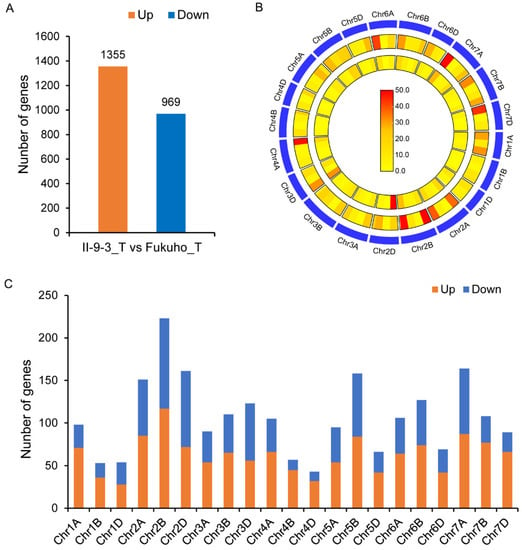
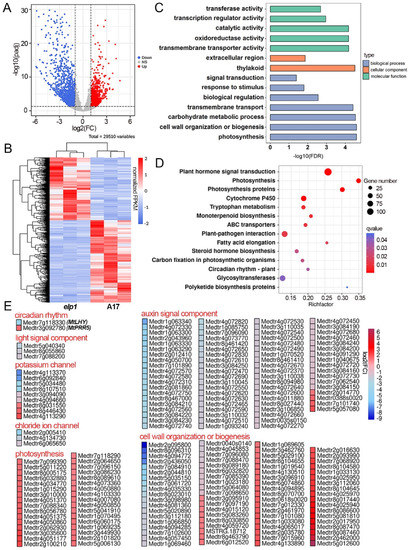
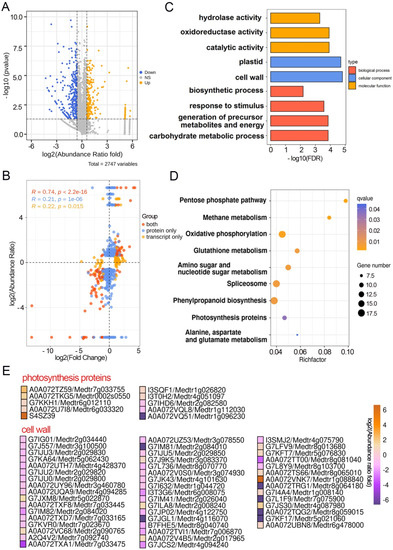
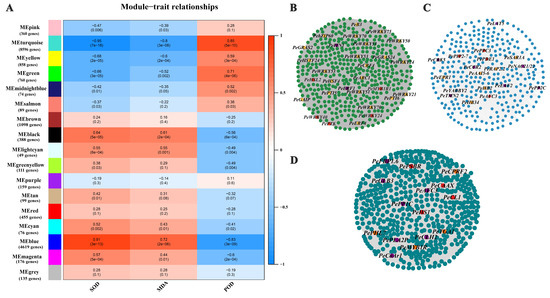
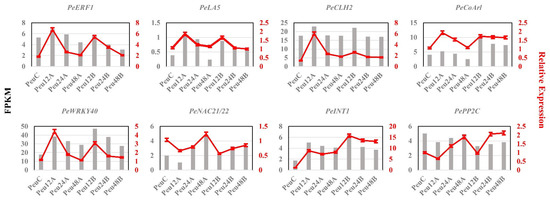
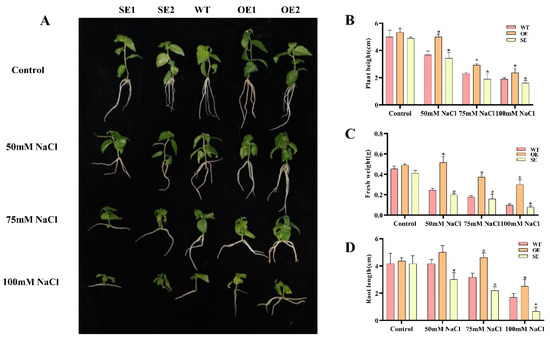
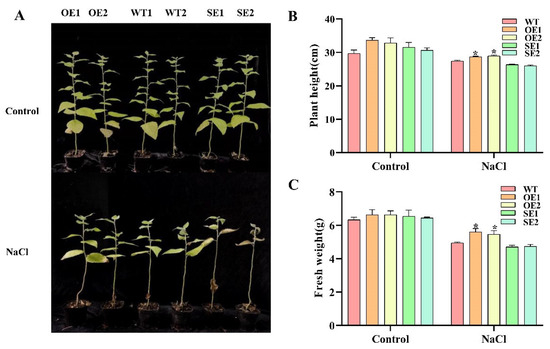
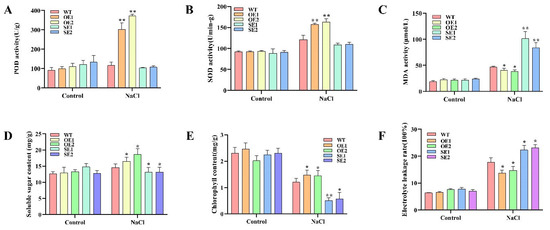
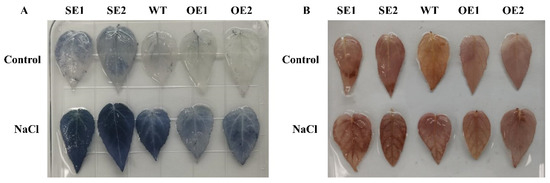
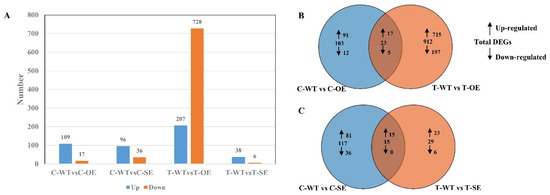
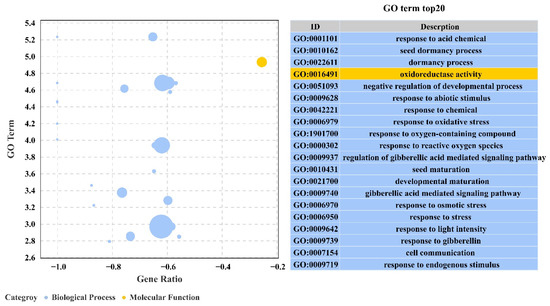
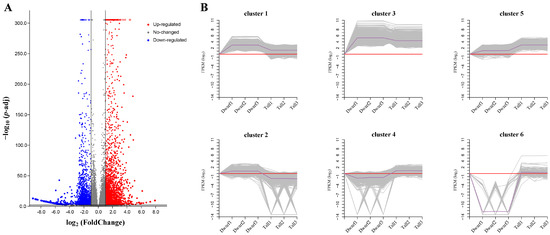
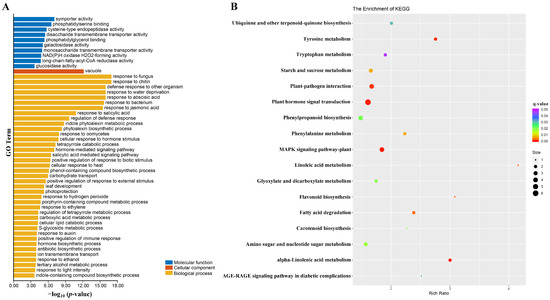
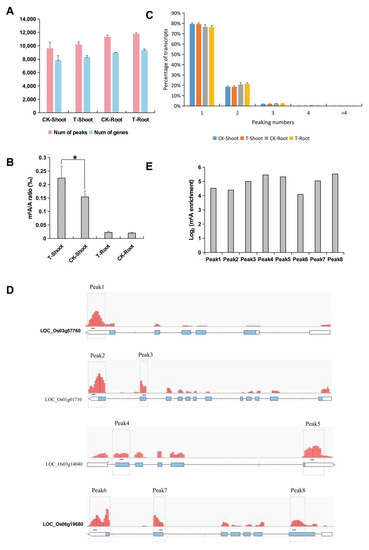
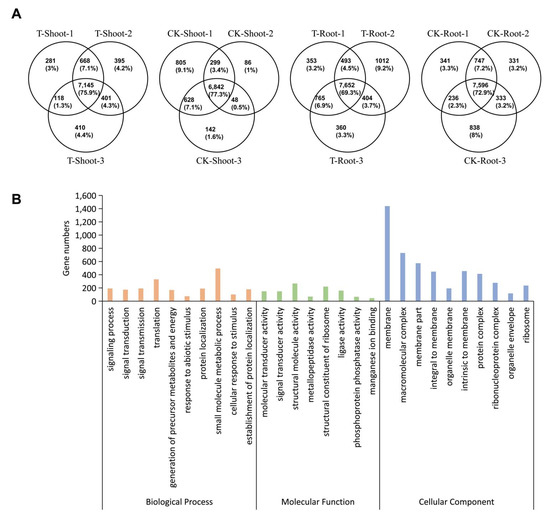
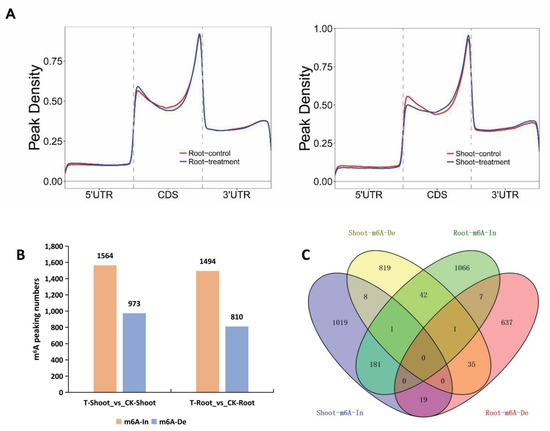
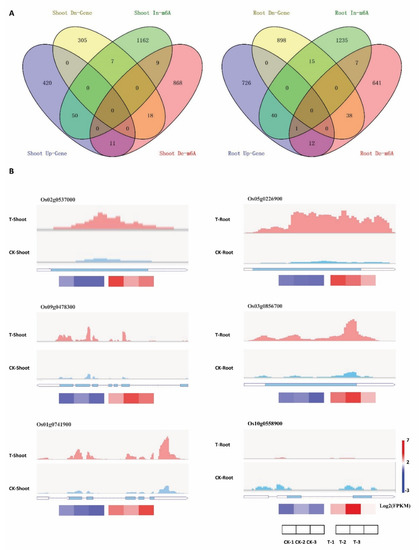
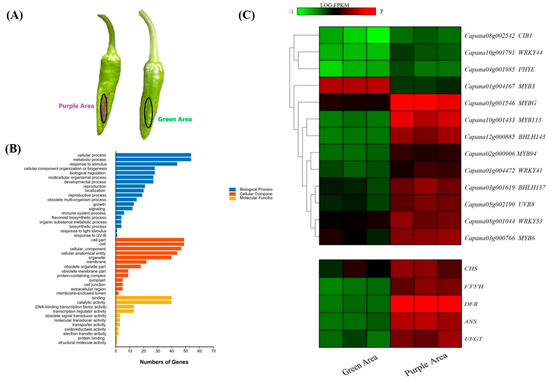
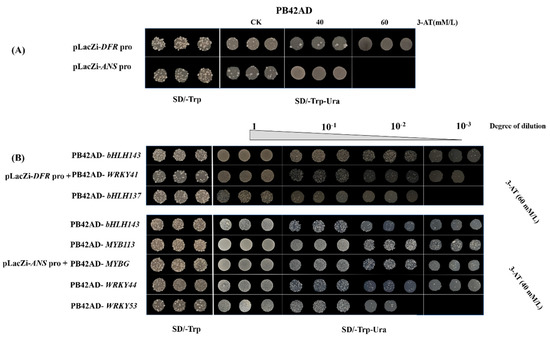
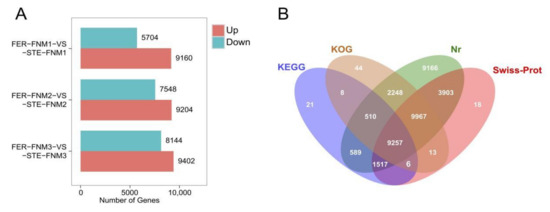
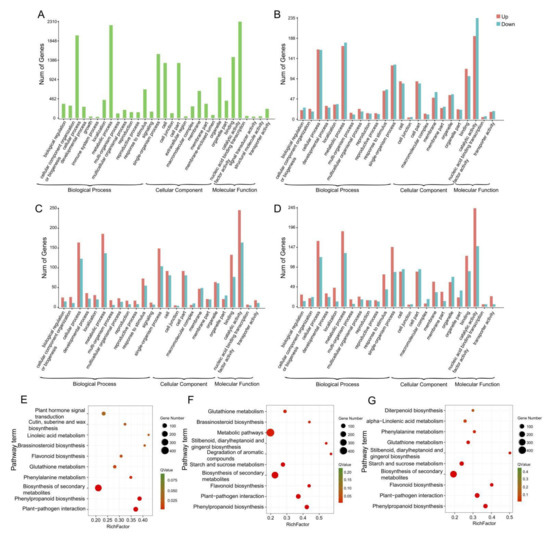
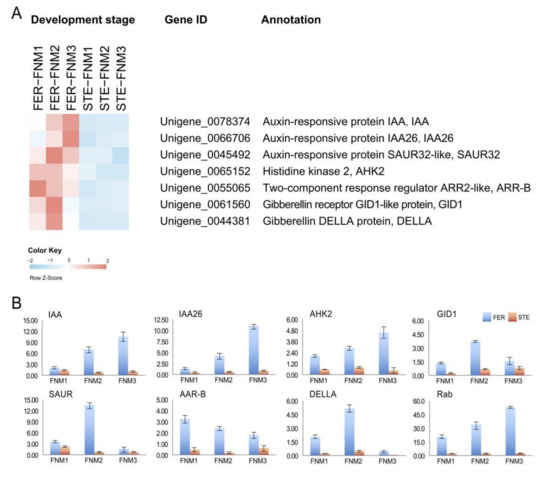
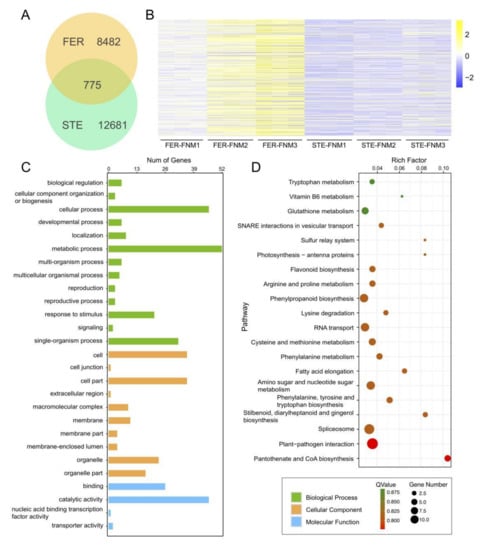
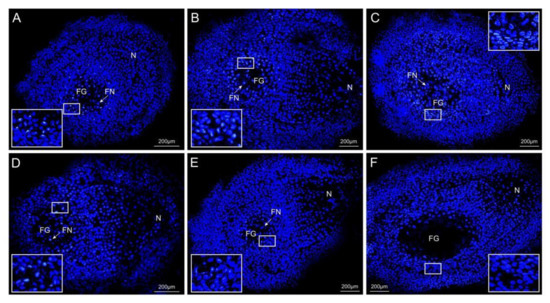
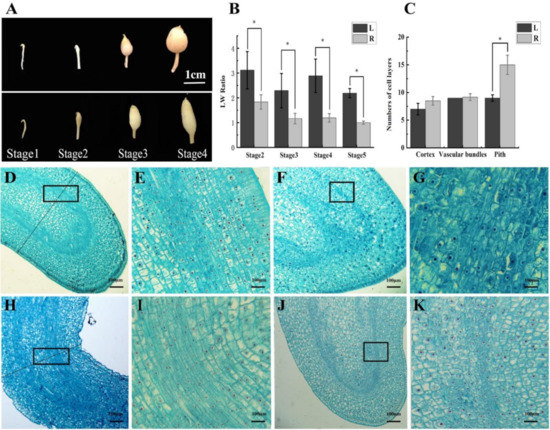
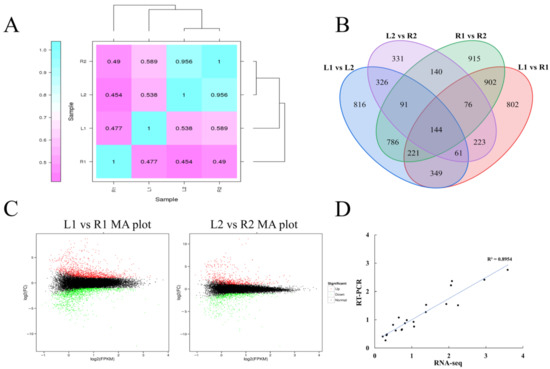
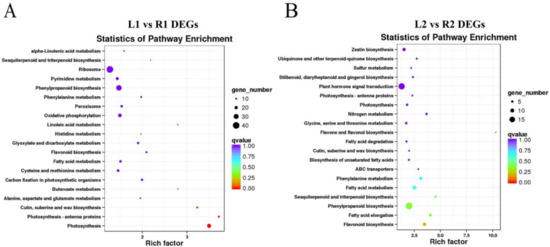
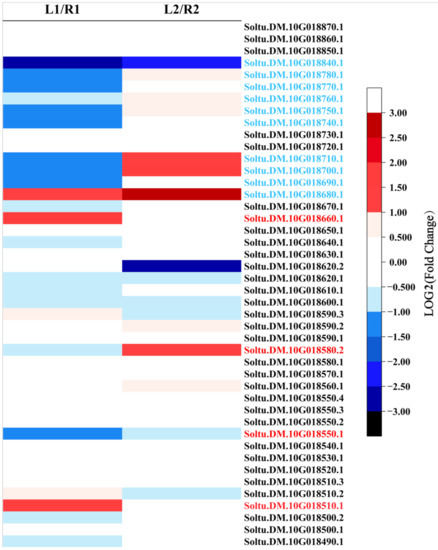
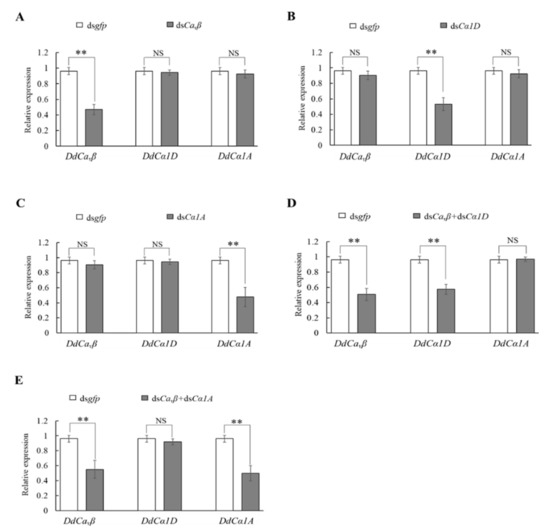
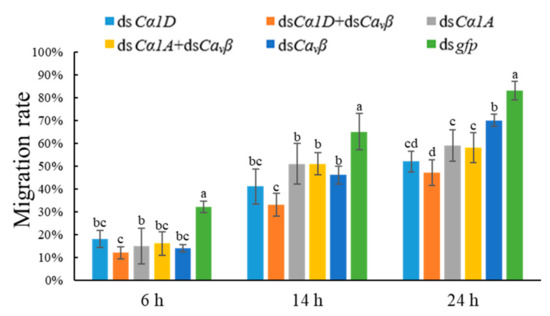
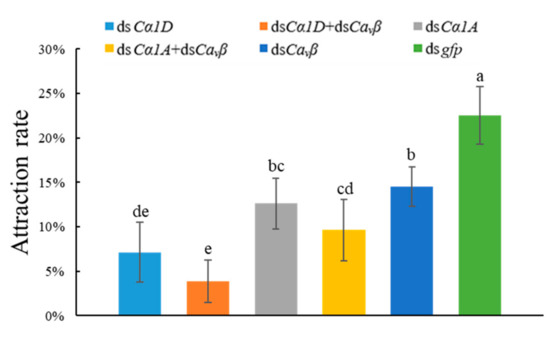
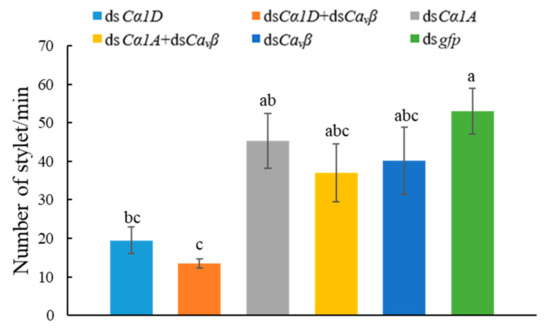
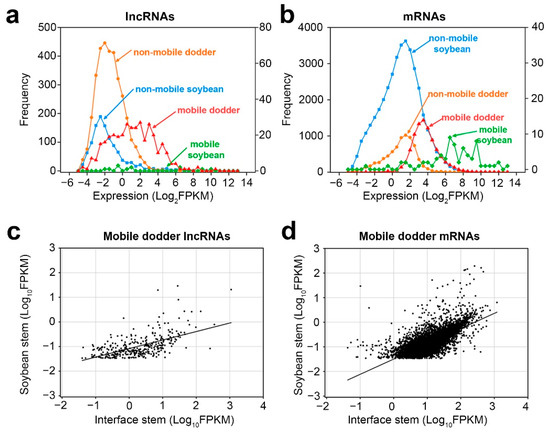
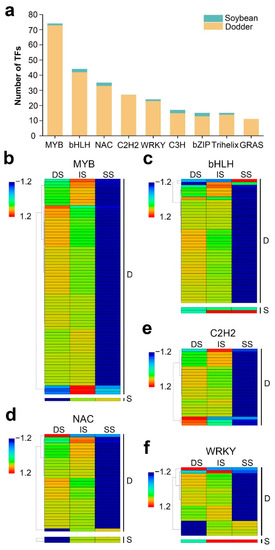
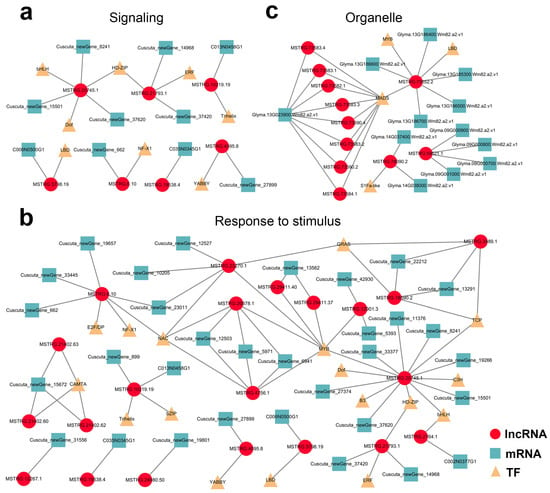
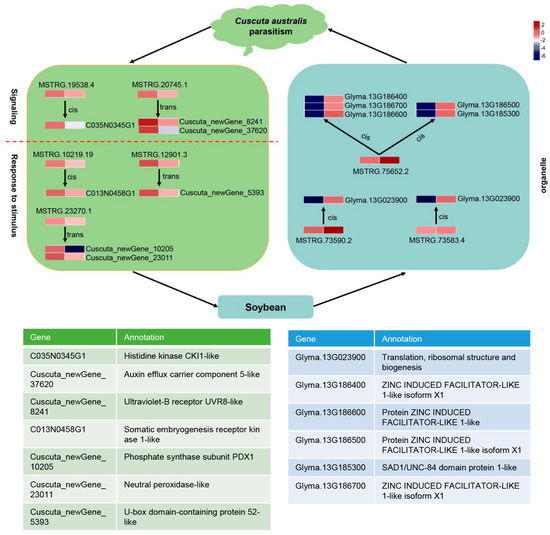
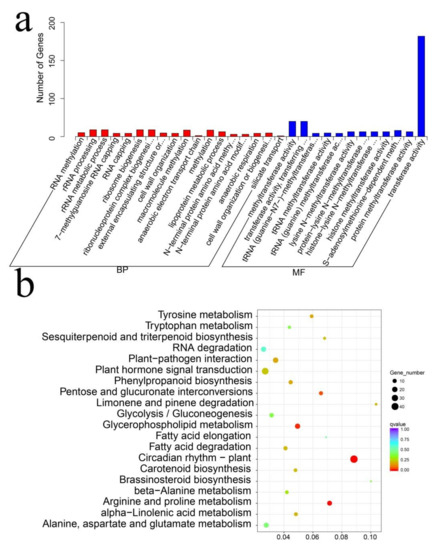
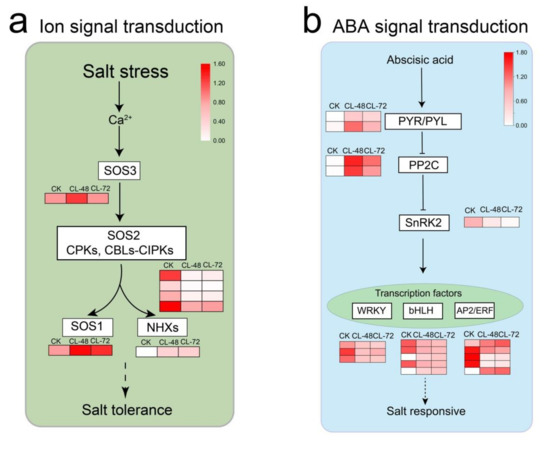
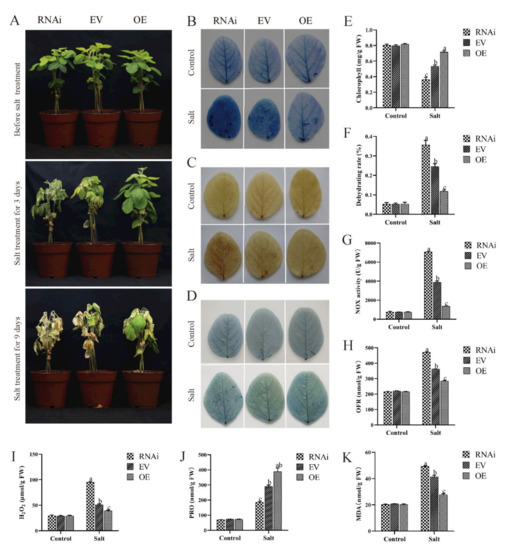
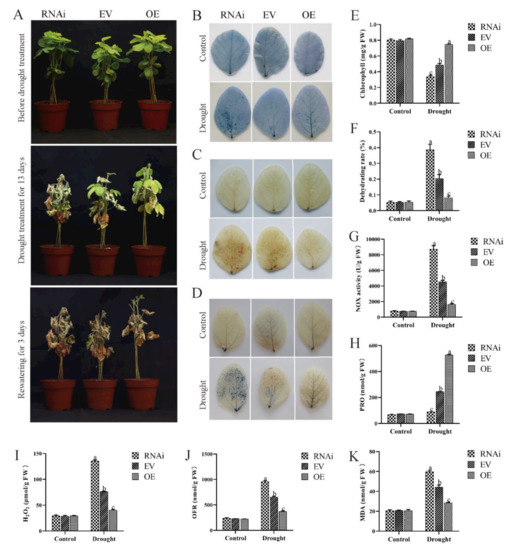
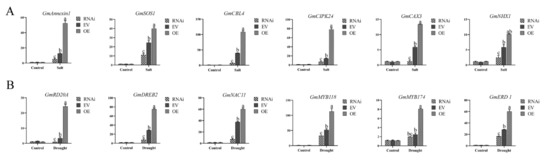
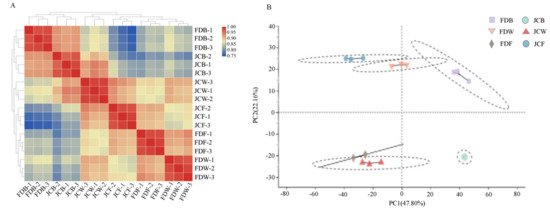
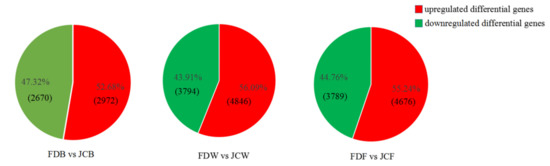
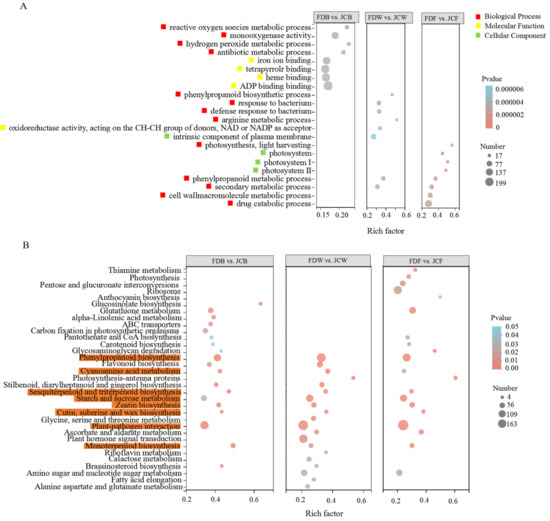
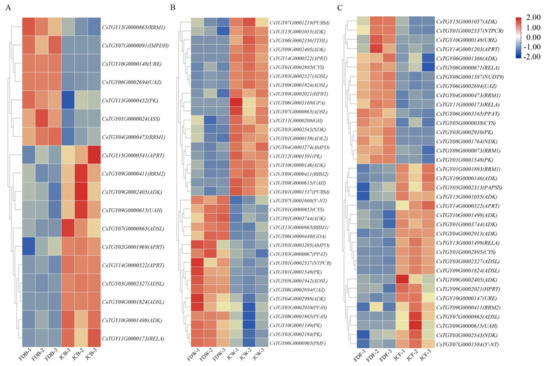
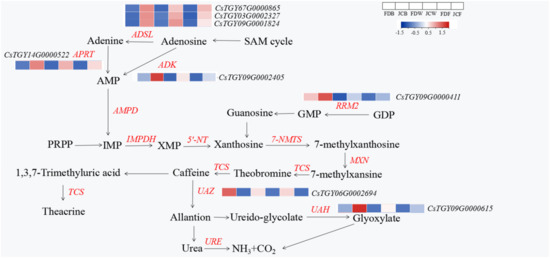
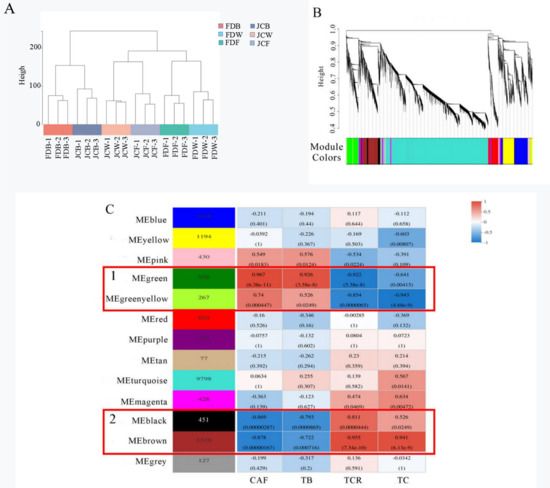
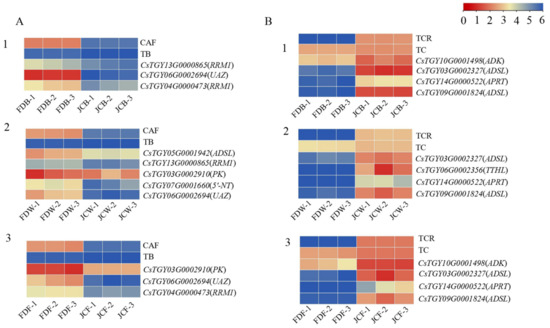
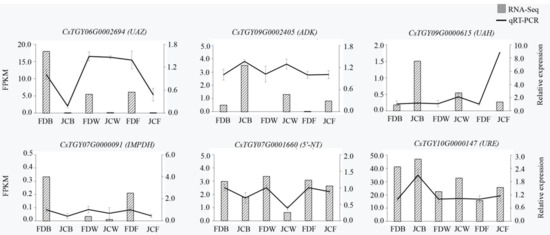
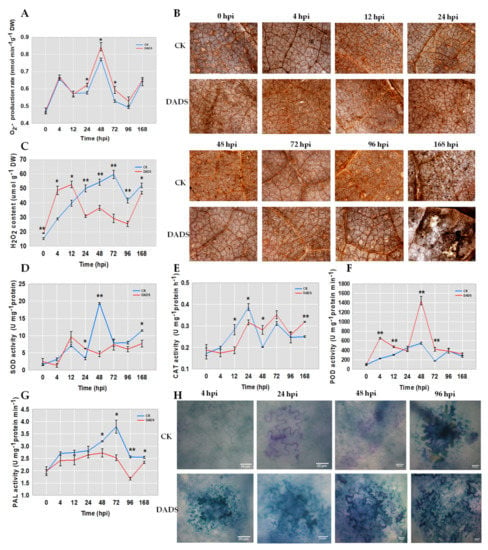
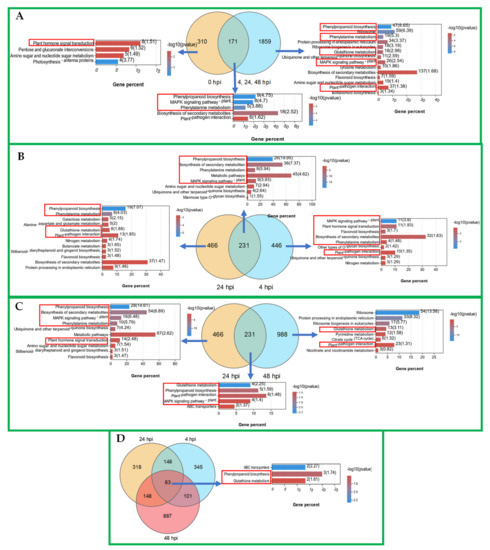
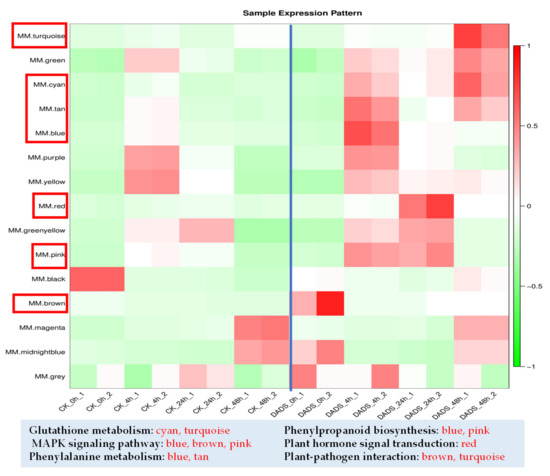
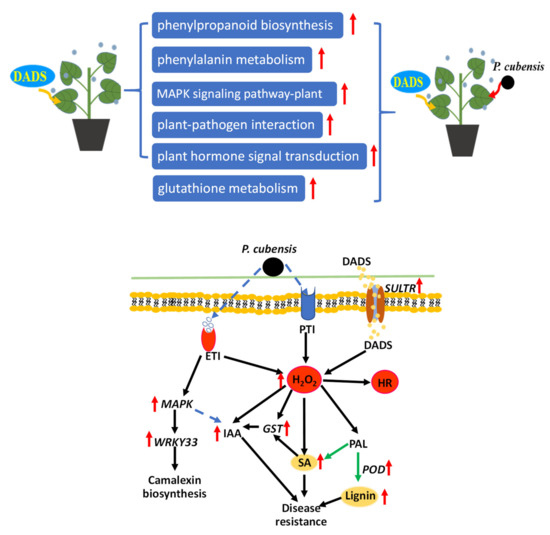
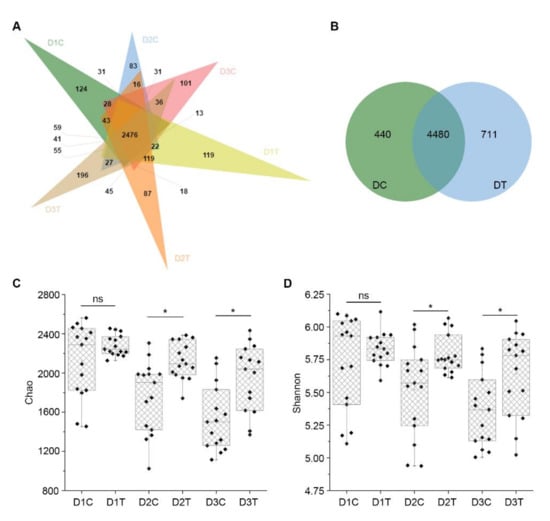
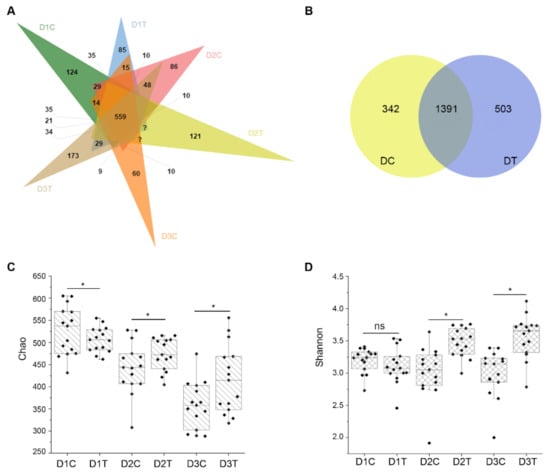
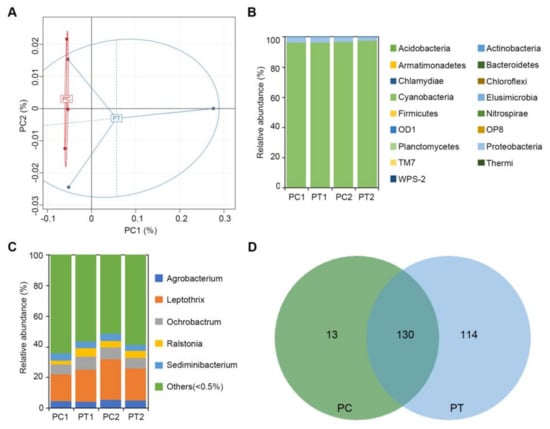
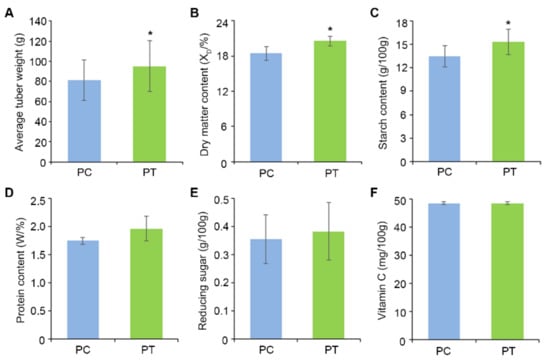
Wheat leaf rust (caused by Puccinia triticina Erikss.) is among the major diseases of common wheat. The lack of resistance genes to leaf rust has limited the development of wheat cultivars. Wheat–Agropyron cristatum (A. cristatum) 2P addition line II-9-3 has been shown to provide broad-spectrum immunity to leaf rust. To identify the specific A. cristatum resistance genes and related regulatory pathways in II-9-3, we conducted a comparative transcriptome analysis of inoculated and uninoculated leaves of the resistant addition line II-9-3 and the susceptible cultivar Fukuhokomugi (Fukuho). The results showed that there were 66 A. cristatum differentially expressed genes (DEGs) and 1389 wheat DEGs in II-9-3 during P. triticina infection. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment and gene set enrichment analysis (GSEA) revealed that the DEGs of II-9-3 were associated with plant–pathogen interaction, MAPK signaling pathway–plant, plant hormone signal transduction, glutathione metabolism, and phenylpropanoid biosynthesis. Furthermore, many defense-related A. cristatum genes, such as two NLR genes, seven receptor kinase-encoding genes, and four transcription factor-encoding genes, were identified. Our results indicated that the key step of resistance to leaf rust involves, firstly, the gene expression of chromosome 2P upstream of the immune pathway and, secondly, the effect of chromosome 2P on the co-expression of wheat genes in II-9-3. The disease resistance regulatory pathways and related genes in the addition line II-9-3 thus could play a critical role in the effective utilization of innovative resources for leaf rust resistance in wheat breeding.
Full article
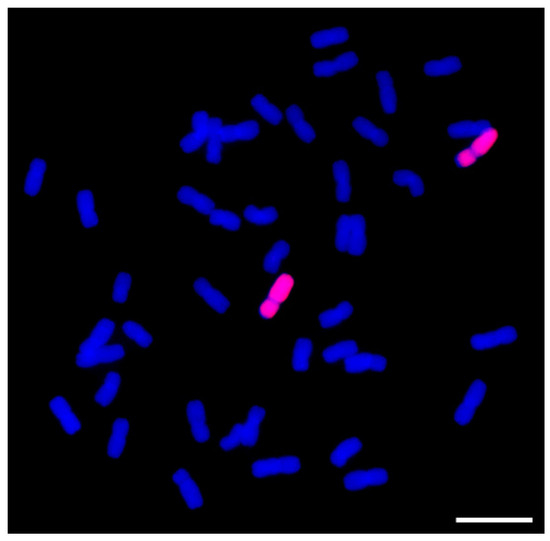
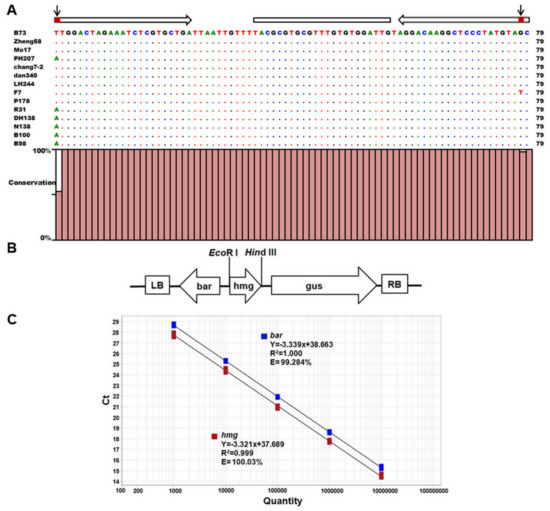
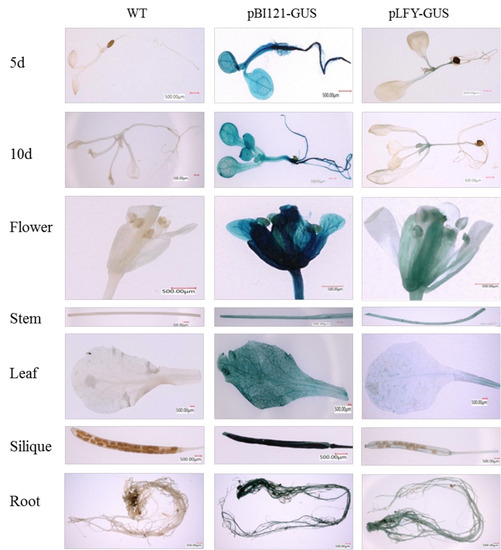
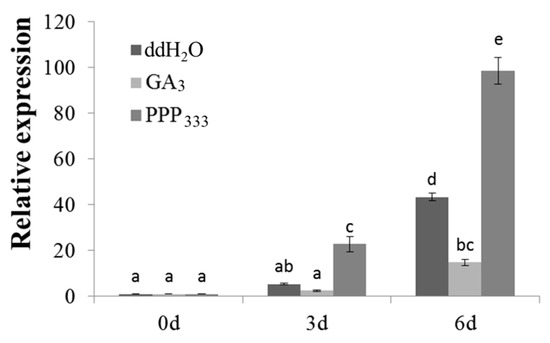
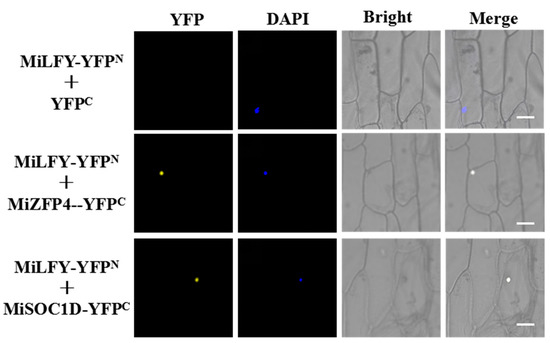
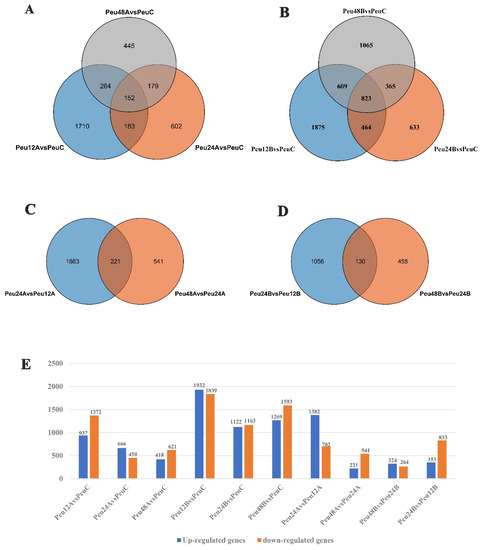
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}