
Global N6-Methyladenosine Profiling Revealed the Tissue-Specific Epitranscriptomic Regulation of Rice Responses to Salt Stress



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
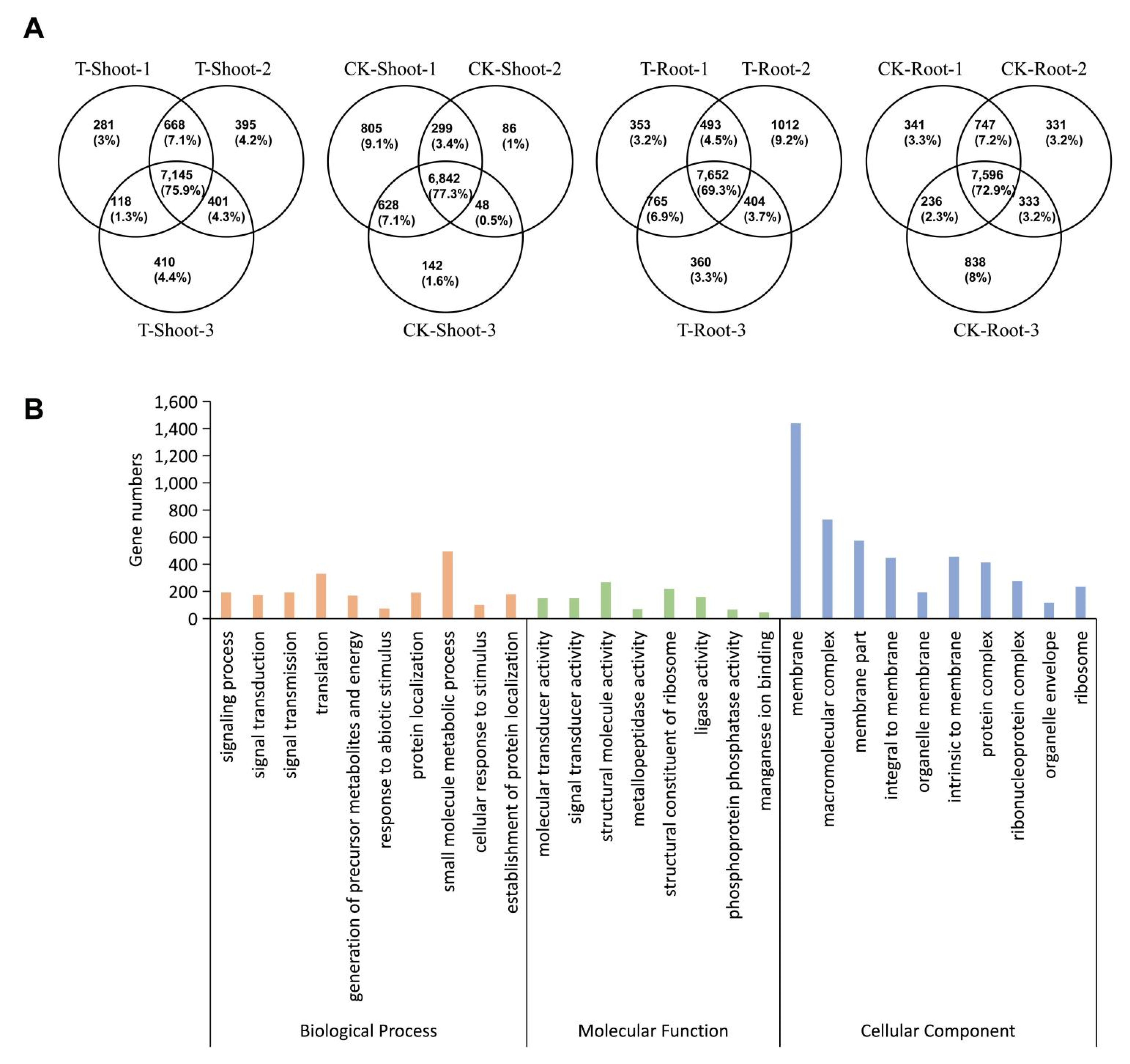
2. Results
2.1. Physiological Indices of FL478 under Salt Stress
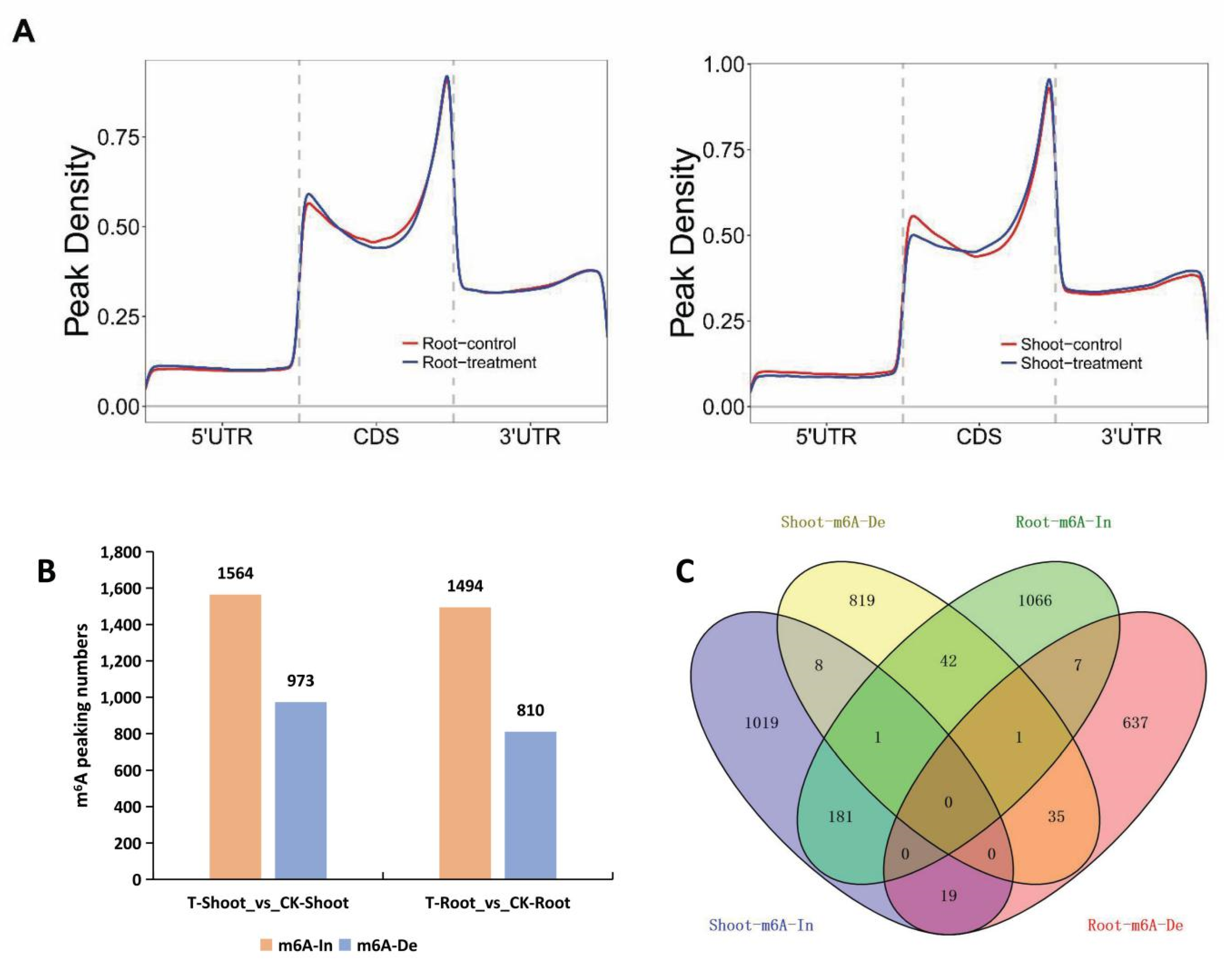
2.2. Identification and Analysis of m6A Methylation Sites
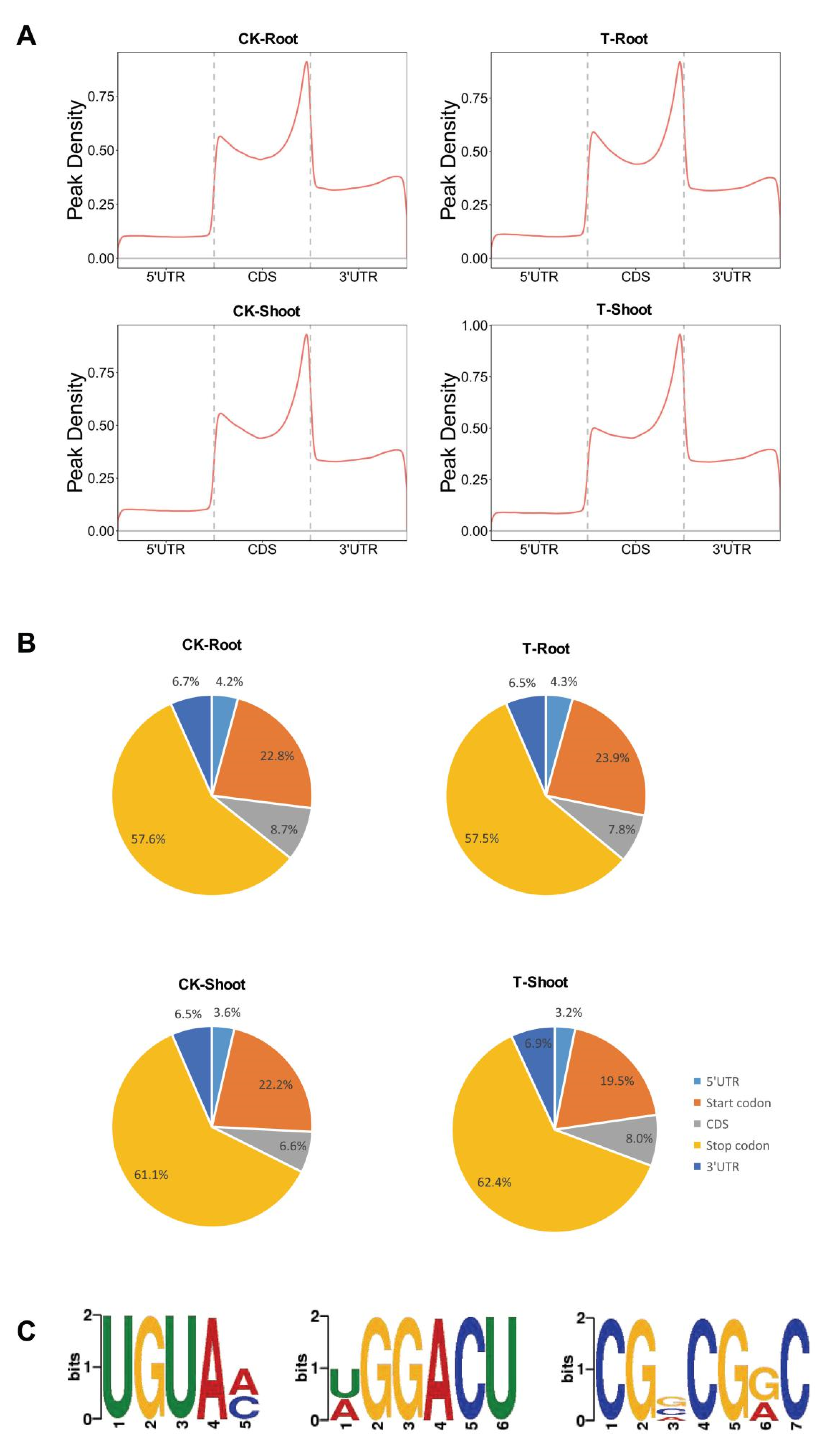
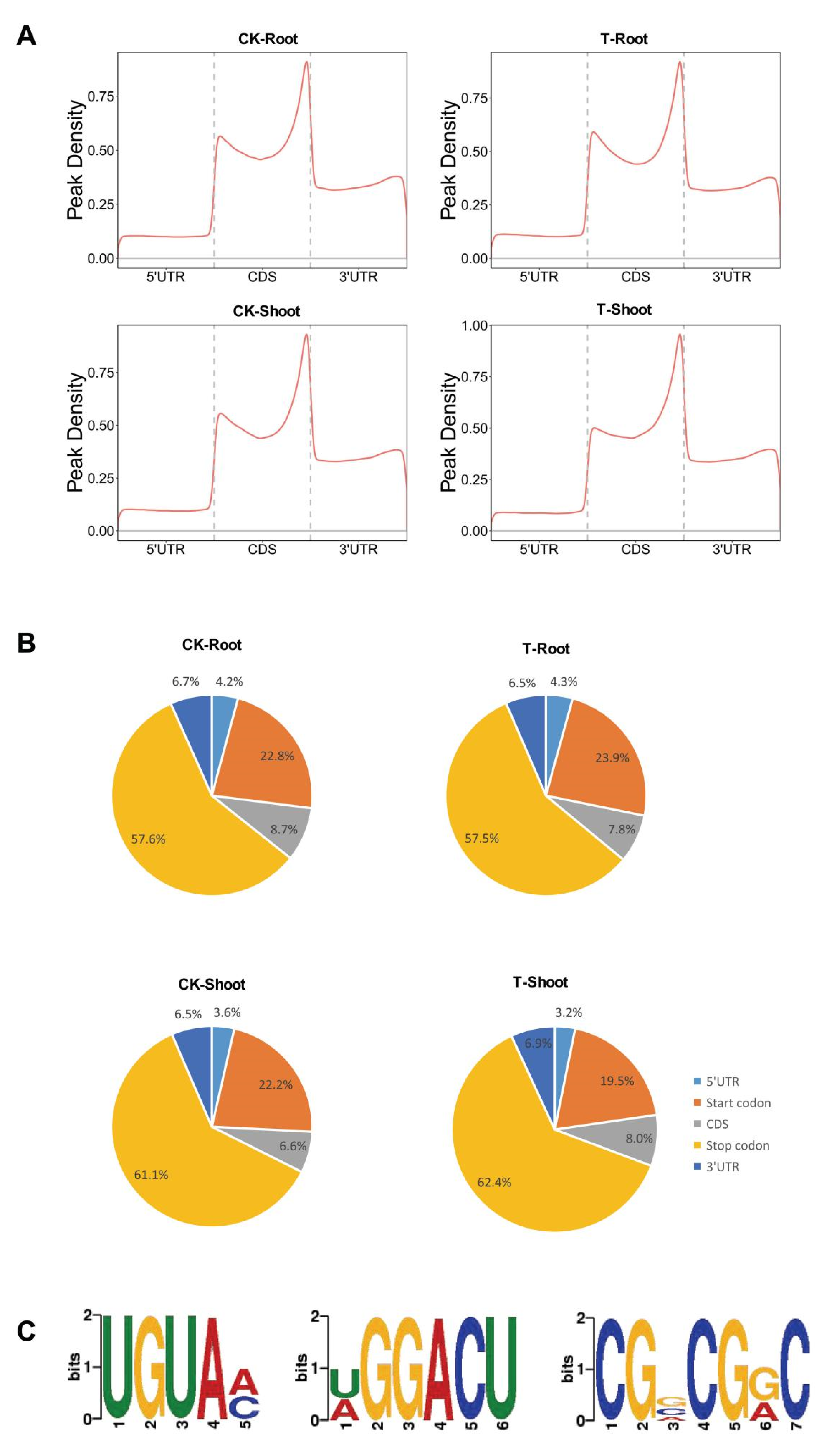
2.3. m6A Distribution among Sequence Motifs in the Transcriptome of FL478
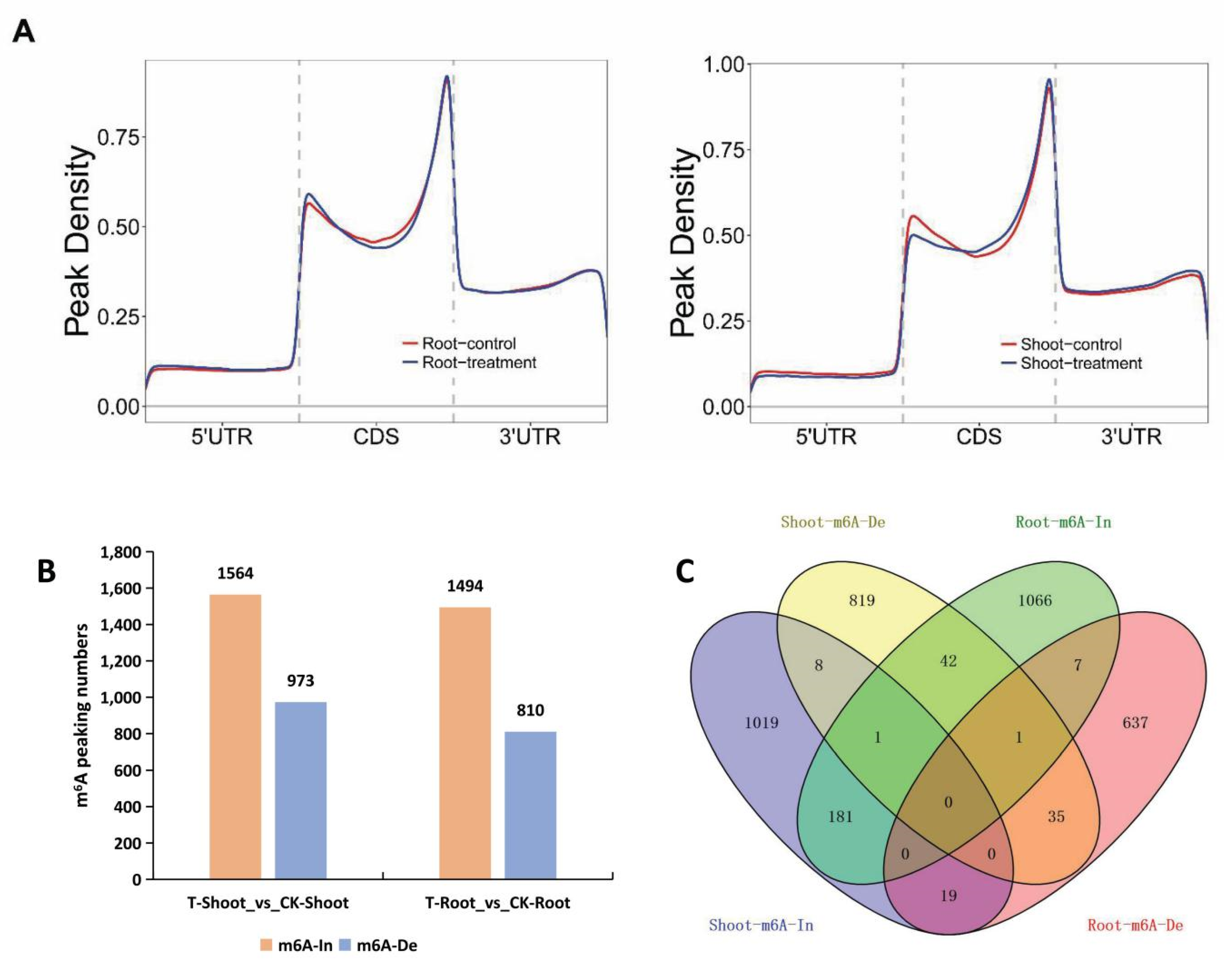
2.4. Tissue-Specific Changes to m6A Methylation in Response to Salt Stress
2.5. A Unique Set of Genes with Tissue-Specific m6A Changes Contributed to Salt Tolerance
2.6. Main Effects of m6A Methylation in Rice Responses to Salt Stress
2.7. m6A Modifications Involved in the Regulation of Gene Expression in FL478 under Salt Stress Conditions
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Analysis of Physiological Indices
4.3. RNA Sequencing (RNA-Seq) and Data Analysis
4.4. MeRIP-Seq Analysis
4.5. Analysis of MeRIP-Seq Data by Quantitative Real-Time (qRT)-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sharp, P.A. The Centrality of RNA. Cell 2009, 136, 577–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arribas-Hernandez, L.; Brodersen, P. Occurrence and functions of m6A and other covalent modifications in plant mRNA. Plant Physiol. 2020, 182, 79–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, H.; Nie, X.J.; Yan, Z.G.; Weining, S. N6-methyladenosine regulatory machinery in plants: Composition, function and evolution. Plant Biotechnol. J. 2019, 17, 1194–1208. [Google Scholar] [CrossRef]
- Shen, L.S.; Liang, Z.; Gu, X.F.; Chen, Y.; Teo, Z.W.; Hou, X.; Cai, W.M.; Dedon, P.C.; Liu, L.; Yu, H. N6-methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev. Cell 2016, 38, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Dominissini, D.; Rechavi, G.; He, C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat. Rev. Genet. 2014, 15, 293–306. [Google Scholar] [CrossRef]
- Li, Y.L.; Wang, X.L.; Li, C.P.; Hu, S.; Yu, J.; Song, S. Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 2014, 11, 1180–1188. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.Z.; MacQueen, A.; Zheng, G.Q.; Duan, H.; Dore, L.C.; Lu, Z.; Liu, J.; Chen, K.; Jia, G.; Bergelson, J.; et al. Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 2014, 5, 5630. [Google Scholar] [CrossRef] [Green Version]
- Duan, H.C.; Wei, L.H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B is an RNA N6-methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell 2017, 29, 2995–3011. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.Z.; Manduzio, S.; Kang, H.S. Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 2019, 10, 500. [Google Scholar] [CrossRef] [Green Version]
- Scutenaire, J.; Deragon, J.M.; Jean, V.; Benhamed, M.; Raynaud, C.; Favory, J.J.; Merret, R.; Bousquet-Antonelli, C. The YTH domain protein ECT2 is an m6A reader required for normal trichome branching in Arabidopsis. Plant Cell 2018, 30, 986–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.H.; Song, P.Z.; Wang, Y.; Lu, Z.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.C.; Jia, G. The m6A reader ECT2 controls trichome morphology by affecting mRNA stability in Arabidopsis. Plant Cell 2018, 30, 968–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huong, T.T.; Ngoc, L.N.T.; Kang, H. Functional characterization of a putative RNA demethylase ALKBH6 in Arabidopsis growth and abiotic stress responses. Int. J. Mol. Sci. 2020, 21, 6707. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Zhang, Y.; He, Q.Z.; Qi, Z.; Zhang, G.; Xu, W.; Yi, T.; Wu, G.; Li, R. MTA, an RNA m6A methyltransferase, enhances drought tolerance by regulating the development of trichomes and roots in poplar. Int. J. Mol. Sci. 2020, 21, 2462. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.J.; Kramer, M.C.; Gosai, S.J.; Yu, X.; Vandivier, L.E.; Nelson, A.; Anderson, Z.D.; Beilstein, M.A.; Fray, R.G.; Lyons, E.; et al. N6-methyladenosine inhibits local ribonucleolytic cleavage to stabilize mRNAs in Arabidopsis. Cell Rep. 2018, 25, 1146–1157. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.Y.; Xu, T.; Kang, H. N6-methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775. [Google Scholar] [CrossRef]
- Miao, Z.Y.; Zhang, T.; Qi, Y.H.; Song, J.; Han, Z.; Ma, C. Evolution of the RNA N6-methyladenosine methylome mediated by genomic duplication. Plant Physiol. 2020, 182, 345–360. [Google Scholar] [CrossRef] [Green Version]
- Tack, D.C.; Su, Z.; Yu, Y.; Bevilacqua, P.C.; Assmann, S.M. Tissue-specific changes in the RNA structurome mediate salinity response in Arabidopsis. RNA 2020, 26, 492–511. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.X.; Sun, X.; Li, J.L.; Song, Y.; Song, J.; Wang, F.; Liu, L.; Zhang, X.; Sui, N. Analysis of N6-methyladenosine reveals a new important mechanism regulating the salt tolerance of sweet sorghum. Plant Sci. 2021, 304, 110801. [Google Scholar] [CrossRef] [PubMed]
- Ganie, S.A.; Molla, K.A.; Henry, R.J.; Bhat, K.V.; Mondal, T.K. Advances in understanding salt tolerance in rice. Theor. Appl. Genet. 2019, 132, 851–870. [Google Scholar] [CrossRef]
- Zheng, D.Y.; Wang, L.; Chen, L.F.; Pan, X.; Lin, K.; Fang, Y.; Wang, X.E.; Zhang, W. Salt-responsive genes are differentially regulated at the chromatin levels between seedlings and roots in Rice. Plant Cell Physiol. 2019, 60, 1790–1803. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.S.; Zhao, X.Q.; Pan, Y.J.; Zhu, L.; Fu, B.; Li, Z. DNA methylation changes detected by methylation-sensitive amplified polymorphism in two contrasting rice genotypes under salt stress. J. Genet. Genom. 2011, 38, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.L.; Tian, S.P.; Qin, G.Z. RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019, 20, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic modifications of mRNA and DNA in plants. Mol. Plant 2020, 13, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Saletore, Y.; Meyer, K.; Korlach, J.; Vilfan, I.D.; Jaffrey, S.; Mason, C.E. The birth of the Epitranscriptome: Deciphering the function of RNA modifications. Genome Biol. 2012, 13, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, K.D.; Jaffrey, S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014, 15, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.; Lee, D.K.; Choi, Y.D.; Kim, J.K. OsIAA6, a member of the rice Aux/IAA gene family, is involved in drought tolerance and tiller outgrowth. Plant Sci. 2015, 236, 304–312. [Google Scholar] [CrossRef]
- Hwang, S.G.; Lee, C.Y.; Tseng, C.S. Heterologous expression of rice 9-cis-epoxycarotenoid dioxygenase 4 (OsNCED4) in Arabidopsis confers sugar oversensitivity and drought tolerance. Bot. Stud. 2018, 59, 2. [Google Scholar] [CrossRef]
- Wang, L.; Guo, K.; Li, Y.; Tu, Y.; Hu, H.; Wang, B.; Cui, X.; Peng, L. Expression profiling and integrative analysis of the CESA/CSL superfamily in rice. BMC Plant Biol. 2010, 10, 282. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, C.; Lin, J.; Liu, J.; Liu, B.; Wang, J.; Huang, A.; Li, H.; Zhao, T. OsMPH1 regulates plant height and improves grain yield in rice. PLoS ONE 2017, 12, e0180825. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Xiong, L. Characterization of a purine permease family gene OsPUP7 involved in growth and development control in rice. J. Integr. Plant Biol. 2013, 55, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Nagano, M.; Jin, S.; Miyagi, A.; Yamaguchi, M.; Kawai-Yamada, M.; Ishikawa, T. Plant-unique cis/trans isomerism of long-chain base unsaturation is selectively required for aluminum tolerance resulting from glucosylceramide-dependent plasma membrane fluidity. Plants 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-throughput deep m(6)A-seq reveals unique differential m(6)A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- Sharoni, A.M.; Nuruzzaman, M.; Satoh, K.; Shimizu, T.; Kondoh, H.; Sasaya, T.; Choi, I.R.; Omura, T.; Kikuchi, S. Gene structures, classification and expression models of the AP2/EREBP transcription factor family in rice. Plant Cell Physiol. 2011, 52, 344–360. [Google Scholar] [CrossRef] [PubMed]
- Abiri, R.; Shaharuddin, N.A.; Maziah, M.; Yusof, Z.N.B.; Atabaki, N.; Sahebi, M.; Valdiani, A.; Kalhori, N.; Azizi, P.; Hanafi, M.M. Role of ethylene and the APETALA 2/ethylene response factor superfamily in rice under various abiotic and biotic stress conditions. Environ. Exp. Bot. 2017, 134, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Viana, V.E.; Busanello, C.; da Maia, L.C.; Pegoraro, C.; Costa de Oliveira, A. Activation of rice WRKY transcription factors: An army of stress fighting soldiers? Curr. Opin. Plant Biol. 2018, 45, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Du, H.; Tang, N.; Li, X.; Xiong, L. Identification and expression profiling analysis of TIFY family genes involved in stress and phytohormone responses in rice. Plant Mol. Biol. 2009, 71, 291–305. [Google Scholar] [CrossRef]
- Zhang, Y.P.; Zhou, J.H.; Wang, L. Mini review roles of the bZIP gene family in rice. Genet. Mol. Res. 2014, 13, 3025–3036. [Google Scholar] [CrossRef]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2020, 48, 909–930. [Google Scholar] [CrossRef]
- Naser, V.; Shani, E. Auxin response under osmotic stress. Plant Mol. Biol. 2016, 91, 661–672. [Google Scholar] [CrossRef]
- Jain, M.; Khurana, J.P. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009, 276, 3148–3162. [Google Scholar] [CrossRef] [PubMed]
- Sledz, P.; Jinek, M. Structural insights into the molecular mechanism of the m6A writer complex. eLife 2016, 5, e18438. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, J.; Xue, Y.; Guan, Z.; Zhang, D.; Liu, Z.; Gong, Z.; Wang, Q.; Huang, J.; Tang, C. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 2016, 534, 260. [Google Scholar] [CrossRef] [PubMed]
- Niessen, M.; Schneiter, R.; Nothiger, R. Molecular identification of virilizer, a gene required for the expression of the sex-determining gene sex-lethal in Drosophila melanogaster. Genetics 2001, 157, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.A.; Liu, J.; Cui, X.L.; Cao, J.; Luo, G.; Zhang, Z.; Cheng, T.; Gao, M.; Shu, X.; Ma, H.; et al. VIRMA mediates preferential m6A mRNA methylation in 3’ UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018, 4, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedeles, B.I.; Singh, V.; Delaney, J.C.; Li, D.; Essigmann, J.M. The AlkB family of Fe(II)/alpha-ketoglutarate-dependent dioxygenases: Repairing nucleic acid alkylation damage and beyond. J. Biol. Chem. 2015, 290, 20734–20742. [Google Scholar] [CrossRef] [Green Version]
- Alemu, E.A.; He, C.; Klungland, A. ALKBHs-facilitated RNA modifications and de-modifications. DNA Repair 2016, 44, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef]
- Welsch, R.; Wüst, F.; Bär, C.; Al-Babili, S.; Beyer, P. A third phytoene synthase is devoted to abiotic stress-induced abscisic acid formation in rice and defines functional diversification of phytoene synthase genes. Plant Physiol. 2008, 147, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.S.; Zhu, H.B.; Jin, G.L.; Liu, H.L.; Wu, W.R.; Zhu, J. Genome-scale identification and analysis of LEA genes in rice (Oryza sativa L.). Plant Sci. 2007, 172, 414–420. [Google Scholar] [CrossRef]
- Cai, H.; Huang, S.; Che, J.; Yamaji, N.; Ma, J.F. The tonoplast-localized transporter OsHMA3 plays an important role in maintaining Zn homeostasis in rice. J. Exp. Bot. 2019, 70, 2717–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Shabala, S.; Shabala, L.; Zhou, M.; Meinke, H.; Venkataraman, G.; Chen, Z.; Zeng, F.; Zhao, Q. Tissue-specific regulation of Na+ and K+ transporters explains genotypic differences in salinity stress tolerance in rice. Front. Plant Sci. 2019, 10, 1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Hong, Y.; Zhu, G.; Li, Y.; Niu, Q.; Yao, J.; Hua, K.; Bai, J.; Zhu, Y.; Shi, H.; et al. Loss of salt tolerance during tomato domestication conferred by variation in a Na+/K+ transporter. EMBO J. 2020, 39, e103256. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Mosa, K.A.; Chhikara, S.; Musante, C.; White, J.C.; Dhankher, O.P. Two rice plasma membrane intrinsic proteins, OsPIP2;4 and OsPIP2;7, are involved in transport and providing tolerance to boron toxicity. Planta 2014, 239, 187–198. [Google Scholar] [CrossRef]
- Yoshida, S.; Forno, D.A.; Cock, J.H.; Gomez, K.A. Routine procedure for growing rice plants in culture solution. In Laboratory Manual for Physiological Studies of Rice; The International Rice Research Institute: Los Baños, Philippines, 1976; pp. 61–66. [Google Scholar]
- Wang, Y.; Du, F.; Wang, J.; Li, Y.; Zhang, Y.; Zhao, X.; Zheng, T.; Li, Z.; Xu, J.; Wang, W.; et al. Molecular dissection of the gene OsGA2ox8 conferring osmotic stress tolerance in rice. Int. J. Mol. Sci. 2021, 22, 9107. [Google Scholar] [CrossRef]
- Song, S.Y.; Chen, Y.; Chen, J.; Dai, X.Y.; Zhang, W.H. Physiological mechanisms underlying OsNAC5-dependent tolerance of rice plants to abiotic stress. Planta 2011, 234, 331–345. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Ouyang, S.Q.; Liu, Y.F.; Liu, P.; Lei, G.; He, S.J.; Ma, B.; Zhang, W.K.; Zhang, J.S.; Chen, S.Y. Receptor-like kinase OsSIK1 improves drought and salt stress tolerance in rice (Oryza sativa) plants. Plant J. 2010, 62, 316–329. [Google Scholar] [CrossRef]
- Arnon, D.I. Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol. 1949, 24, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 2226–6089. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Du, F.; Li, Y.; Wang, J.; Zhao, X.; Li, Z.; Xu, J.; Wang, W.; Fu, B. Global N6-Methyladenosine Profiling Revealed the Tissue-Specific Epitranscriptomic Regulation of Rice Responses to Salt Stress. Int. J. Mol. Sci. 2022, 23, 2091. https://doi.org/10.3390/ijms23042091
Wang Y, Du F, Li Y, Wang J, Zhao X, Li Z, Xu J, Wang W, Fu B. Global N6-Methyladenosine Profiling Revealed the Tissue-Specific Epitranscriptomic Regulation of Rice Responses to Salt Stress. International Journal of Molecular Sciences. 2022; 23(4):2091. https://doi.org/10.3390/ijms23042091
Chicago/Turabian StyleWang, Yinxiao, Fengping Du, Yingbo Li, Juan Wang, Xiuqin Zhao, Zhikang Li, Jianlong Xu, Wensheng Wang, and Binying Fu. 2022. "Global N6-Methyladenosine Profiling Revealed the Tissue-Specific Epitranscriptomic Regulation of Rice Responses to Salt Stress" International Journal of Molecular Sciences 23, no. 4: 2091. https://doi.org/10.3390/ijms23042091
APA StyleWang, Y., Du, F., Li, Y., Wang, J., Zhao, X., Li, Z., Xu, J., Wang, W., & Fu, B. (2022). Global N6-Methyladenosine Profiling Revealed the Tissue-Specific Epitranscriptomic Regulation of Rice Responses to Salt Stress. International Journal of Molecular Sciences, 23(4), 2091. https://doi.org/10.3390/ijms23042091