
High-Throughput and Accurate Determination of Transgene Copy Number and Zygosity in Transgenic Maize: From DNA Extraction to Data Analysis


Abstract
1. Introduction
2. Results
2.1. Discovered SNPs in Target Regions of Endogenous Reference Gene among Maize Cultivars
2.2. SNPs of Endogenous Reference Targets Affect qPCR Efficiency
2.3. Development of Duplexed TaqMan Assay in 384-Well Format
2.4. Reproducibility of TaqMan Copy Number Assay
2.5. Accuracy of TaqMan Copy Number Assay by Southern Blot and dPCR
2.6. Feasibility of One Technical Repetition of TaqMan Assay
2.7. Integrated High-Throughput Platform for Performing DNA Extraction, TaqMan Assay, and Data Analysis
3. Discussion
3.1. Development of High-Throughput Copy Number Assay Using a “Universal” Maize Reference Gene System
3.1.1. Allelic Variation of hmg Gene among Maize Cultivars
3.1.2. Development of Duplexed TaqMan Assay in 384-Well Format for High-Throughput Copy-Number Screening
3.2. High Accuracy of TaqMan Copy Number Assay without Technical Repeats, Comparable to dPCR
3.2.1. High Accuracy of Copy Number Determination by TaqMan Assay Is the Same as dPCR, Higher Than Southern Blot Analysis
3.2.2. High Accuracy of TaqMan Assay Achieved without Technical Repeats
3.3. High-Throughput Analysis of Transgene Copy Number and Zygosity
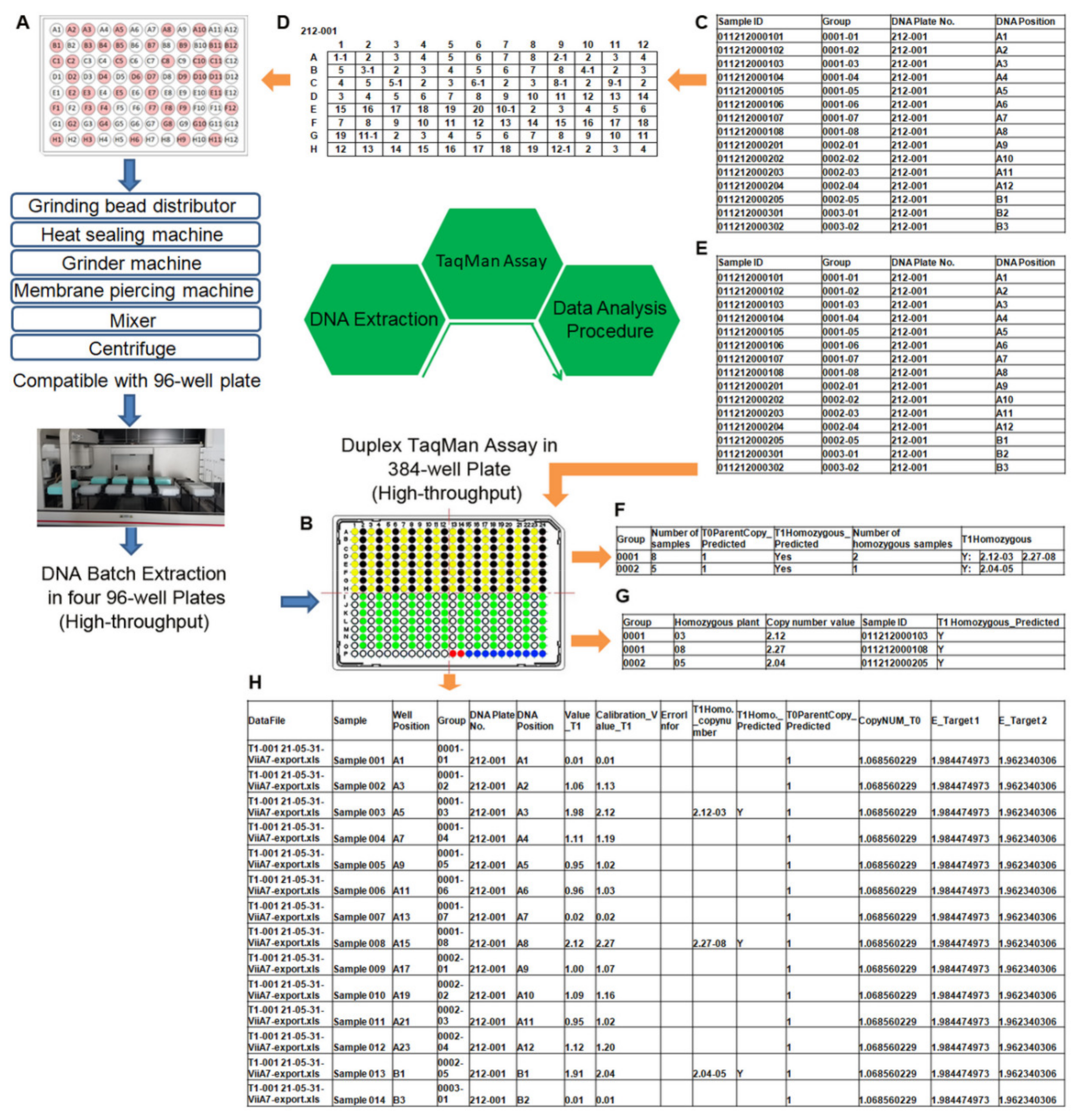
- A suitable range for genomic DNA was 10–400 ng. The multiple time-consuming and laborious manipulation steps in the preparation of high-quality genomic DNA from thousands of samples still remains a primary limitation when thousands of samples need to be analyzed rapidly, such as in molecular plant breeding and plant transformation programs. In addition, having high-quality and intact genomic DNA is an important prerequisite for the accurate measurement of transgene copy numbers. The use of poor-quality genomic DNA produces qPCR results that are not accurate [43]. To overcome these problems, we developed a rapid and high-throughput method for the extraction of high-quality and intact DNA using magnetic beads on a Beckman automated liquid handler (Biomek FXp). The procedure described here was designed and optimized to extract four plates of 96-well DNA at the same time and allow one user to easily extract 28 plates of 96-well DNA within 8 h (see Section 4). Most importantly, the resulting DNA is of excellent quality and suitable for use in transgene copy number and zygosity analysis by the TaqMan assay.
- The uniformity of the endogenous reference gene was our main concern when we were preparing to develop the TaqMan assay for transgene copy numbers. In this study, one previously reported qPCR assay on maize endogenous reference gene hmg was comprehensively evaluated for target gene sequence variations among 633 maize inbred lines and qPCR performance. Two SNPs were observed in the hmg gene, and these SNPs significantly increased the efficiency of qPCR amplification. The new TaqMan assay employs redesigned homogeneous primers and probe, eliminating SNPs, and has been validated to be most useful in establishing accurate and creditable quantitative PCR analysis of transgenic maize.
- A standard reference plasmid was developed to include two target sequences in a single plasmid. In this way, a serial dilution of the dual-target plasmid DNA with correct concentration could be made to obtain accurate standard curves for the exogenous bar and endogenous hmg genes, which is a key prerequisite for quantitative analysis. The strengths of the developed pCAMBIA3301-ZmHMG plasmid are its complete characterization of sequence and structure, high performance, ease of preparation, stability in storage, and low cost. It was previously shown that the use of cloned plasmid GMO target sequences for calibration can produce accurate quantitative results [44,45,46,47].
- A transgenic T0 plant with one copy of the transgene demonstrated by dPCR was applied as an inter-run calibrator for normalization. Inter-run calibration is required because there is a dependent relationship between quantification cycle value and relative quantity due to instrument-related variation (PCR block, lamp, filters, detectors, and so on), data analysis settings (baseline correction and threshold), reagents (polymerase, fluorophores, and so on), and the optical properties of plastics. Therefore, inter-run calibration is required to correct for possible run-to-run variation whenever all samples are not analyzed in the same run [48].
- Duplexed reactions were used, as they are more efficient and economical, appropriate for high throughput, and less prone to technical errors than two independent reactions. They are especially valuable for quantitative experiments that require a higher degree of precision, such as copy number assays. It is important to note that the concentrations of primers and probes of the target bar and reference hmg genes should be optimized to avoid preferential amplification of one of the two, because amplification of both genes was carried out in the same reaction tube.
- The duplexed TaqMan assay was performed in 384-well plates with a volume of 10 µL, providing an eightfold increase in throughput over the conventional uniplex 96-well format assay, and a 16-fold reduction in cost. Although there is an ultra-high-throughput fluorescent quantitative PCR system with a 1536-well block currently on the market, it has to be equipped with an automatic liquid dispensing system to pipette with ultra-micro (0.5–2 μL) volume, preventing its application in most testing and research laboratories. In our lab, with proper training, proper tools (for example, a 12-well multichannel pipette), and a good experimental protocol, any conscientious staff member can pipette reagents into 384-well plates and generate consistent high-quality data without the need for robotics or other automated liquid dispensing systems.
- The transgene copy numbers were estimated by the efficiency-corrected method described by Pfaffl [49]. The comparative 2-ΔΔCt method, which is the most widely used method for copy number determination by qPCR, has an often untrue assumption that both the transgene and reference gene have approximately equal and near 100% efficiency, a condition that is rarely met in practice [8,50]. The Pfaffl method includes the different levels of efficiency of the two genes in the equation, and, therefore, the difference in efficiency between the two genes will be accounted for in the calculation [49]. Considering that there are differences in amplification efficiency of the transgene and the reference gene in most situations, the Pfaffl method was adapted to estimate transgene copy numbers in the present study.
- Once the above points were satisfactorily met, the accuracy of the TaqMan assay could be guaranteed, and, on this basis, only one technical repetition needed to be performed. We performed a simple random sampling analysis on the copy number results of 4409 transgenic plants, and the results showed that the accuracy of one repetition could reach 96.7% of that of three repetitions of single-copy plants. For further confirmation, the copy numbers and separation ratios of T1 and T2 plants of 2016 T0 transformants, which have a single copy determined with one technical repetition, were analyzed. For one copy, the reliability of one replicate reached as high as 95.8%. To the best of our knowledge, this is the first demonstration that the TaqMan copy number assay can have comparable accuracy to dPCR without technical repeats. The application of this approach would reduce the cost by two thirds and increase throughput by two thirds compared with three repetitions. In addition, this approach would make it easier to arrange four 96-well samples in a 384-well qPCR plate and to analyze large amounts of data.
- Accurate analysis of large amounts of data was another feature required by high-throughput fluorescence qPCR technology. In addition to the software that comes with the instrument, which can analyze fluorescence quantitative PCR data, we developed data analysis software to process high-throughput data within a few minutes without being limited by the amount of data.
4. Materials and Methods
4.1. Plant Material
4.2. High-Throughput Genomic DNA Extraction Method
4.3. Sequencing and Alignment of Endogenous Reference Gene Target Region
4.4. Construction of Standard Plasmid as Reference Molecule
4.5. Duplexed TaqMan Assay
4.5.1. Primers and Probes
4.5.2. Optimization of TaqMan Primer Concentrations
4.5.3. Duplexed TaqMan qPCR Reaction
4.5.4. Establishment of Standard Curve
4.5.5. Calculation of Copy Number by Pfaffl Method
4.5.6. Evaluation of the Reproducibility of the TaqMan Assay
4.6. Southern Blot Analysis
4.7. Duplexed Digital PCR Analysis
4.8. Simple Random Sampling Analysis
4.9. Data Analysis Program
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mladenovic-Drinic, S.; Ignjatovic-Micic, D.; Eric, I.; Andjelkovic, V.; Jelovac, D.; Konstantinov, K. Biotechnology in maize breeding. Genetika 2004, 36, 93–109. [Google Scholar] [CrossRef]
- James, C. Global Status of Commercialized Biotech/GM Crops: 2019. ISAAA Brief 55: Executive Summary. Available online: http://www.isaaa.org/resources/publications/briefs/55/executivesummary/default.asp (accessed on 22 September 2021).
- Zale, J. Transgenic Plant Analysis. In Plant Biotechnology and Genetics; Stewart, C.N., Jr., Ed.; Wiley: Hoboken, NJ, USA, 2008; pp. 275–290. [Google Scholar]
- Vaucheret, H.; Béclin, C.; Elmayan, T.; Feuerbach, F.; Godon, C.; Morel, J.-B.; Mourrain, P.; Palauqui, J.-C.; Vernhettes, S. Transgene-induced gene silencing in plants. Plant J. 1998, 16, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P. Stabilities and Instabilities in Transgene Expression. In Transgenic Plant Research; Lindsey, K., Ed.; Harwood Academic: Amsterdam, The Netherlands, 1998; pp. 263–275. [Google Scholar]
- Dai, N.; Schaffer, A.; Petreikov, M.; Shahak, Y.; Giller, Y.; Ratner, K.; Levine, A.; Granot, D. Overexpression of Arabidopsis hexokinase in tomato plants inhibits growth, reduces photosynthesis, and induces rapid senescence. Plant Cell 1999, 11, 1253–1266. [Google Scholar] [CrossRef] [PubMed]
- Passricha, N.; Batra, R.; Behl, R.K.; Sikka, V.K. Differential and temperature dependent regulation of ADP-glucose pyro-phosphorylase by specific chromosome in wheat grains. Cereal Res. Comm. 2015, 43, 591–603. [Google Scholar] [CrossRef][Green Version]
- Bubner, B.; Baldwin, I.T. Use of real-time PCR for determining copy number and zygosity in transgenic plants. Plant Cell Rep. 2004, 23, 263–271. [Google Scholar] [CrossRef]
- Mason, G.; Provero, P.; Vaira, A.M.; Accotto, G.P. Estimating the number of integrations in transformed plants by quantitative real-time PCR. BMC Biotech. 2002, 2, 1–10. [Google Scholar] [CrossRef]
- Ingham, D.J.; Beer, S.; Money, S.; Hansen, G. Quantitative Real-Time PCR Assay for Determining Transgene Copy Number in Transformed Plants. BioTechniques 2001, 31, 132–140. [Google Scholar] [CrossRef]
- Mieog, J.C.; Howitt, C.; Ral, J.-P. Fast-tracking development of homozygous transgenic cereal lines using a simple and highly flexible real-time PCR assay. BMC Plant Biol. 2013, 13, 1–9. [Google Scholar] [CrossRef]
- Bubner, B.; Gase, K.; Baldwin, I.T. Two-fold differences are the detection limit for determining transgene copy numbers in plants by real-time PCR. BMC Biotechnol. 2004, 4, 14. [Google Scholar] [CrossRef][Green Version]
- Gadaleta, A.; Giancaspro, A.; Cardone, M.F.; Blanco, A. Real-time PCR for the detection of precise transgene copy number in durum wheat. Cell. Mol. Biol. Lett. 2011, 16, 652–668. [Google Scholar] [CrossRef]
- Li, Z.; Hansen, J.L.; Liu, Y.; Zemetra, R.S.; Berger, P.H. Using real-time PCR to determine transgene copy number in wheat. Plant Mol. Biol. Rep. 2004, 22, 179–188. [Google Scholar] [CrossRef]
- Shou, H.; Frame, B.R.; Whitham, S.A.; Wang, K. Assessment of transgenic maize events produced by particle bombardment or Agrobacterium-mediated transformation. Mol. Breed. 2004, 13, 201–208. [Google Scholar] [CrossRef]
- Yang, L.; Ding, J.; Zhang, C.; Jia, J.; Weng, H.; Liu, W.; Zhang, D. Estimating the copy number of transgenes in transformed rice by real-time quantitative PCR. Plant Cell Rep. 2005, 23, 759–763. [Google Scholar] [CrossRef]
- Prior, F.A.; Tackaberry, E.S.; Aubin, R.A.; Casley, W.L. Accurate Determination of Zygosity in Transgenic Rice by Real-time PCR Does Not Require Standard Curves or Efficiency Correction. Transgenic Res. 2006, 15, 261–265. [Google Scholar] [CrossRef]
- German, M.; Kandel-Kfir, M.; Swarzberg, D.; Matsevitz, T.; Granot, D. A rapid method for the analysis of zygosity in transgenic plants. Plant Sci. 2003, 164, 183–187. [Google Scholar] [CrossRef]
- Schmidt, M.A.; Parrot, W.A. Quantitative detection of transgenes in soybean (Glycine max L. Merrill) and peanut (Arachis hypogaea L.) by real-time polymerase chain reaction. Plant. Cell Rep. 2001, 20, 422–428. [Google Scholar] [CrossRef]
- Demeke, T.; Dobnik, D. Critical assessment of digital PCR for the detection and quantification of genetically modified organisms. Anal. Bioanal. Chem. 2018, 410, 4039–4050. [Google Scholar] [CrossRef]
- Glowacka, K.; Kromdijk, J.; Leonelli, L.; Niyogi, K.K.; Clemente, T.E.; Long, S.P. An evaluation of new and established methods to determine T-DNA copy number and homozygosity in transgenic plants. Plant Cell Environ. 2016, 39, 908–917. [Google Scholar] [CrossRef]
- Xu, X.; Peng, C.; Wang, X.; Chen, X.; Wang, Q.; Xu, J. Comparison of droplet digital PCR with quantitative real-time PCR for determination of zygosity in transgenic maize. Transgenic Res. 2016, 25, 855–864. [Google Scholar] [CrossRef]
- Collier, R.; Dasgupta, K.; Xing, Y.; Hernandez, B.T.; Shao, M.; Rohozinski, D.; Kovak, E.; Lin, J.; de Oliveira, M.L.P.; Stover, E.; et al. Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J. 2017, 90, 1014–1025. [Google Scholar] [CrossRef]
- Sun, Y.; Aiyar, P. Application of droplet digital PCR to determine copy number of endogenous genes and transgenes in sug-arcane. Plant. Cell Rep. 2017, 36, 1775–1783. [Google Scholar] [CrossRef]
- Morcia, C.; Ghizzoni, R.; Delogu, C.; Andreani, L.; Carnevali, P.; Terzi, V. Digital PCR: What Relevance to Plant Studies? Biology 2020, 9, 433. [Google Scholar] [CrossRef]
- Hernández, M.; Río, A.; Esteve, T.; Prat, S.; Pla, M. A Rapeseed-Specific Gene, Acetyl-CoA Carboxylase, Can Be Used as a Reference for Qualitative and Real-Time Quantitative PCR Detection of Transgenes from Mixed Food Samples. J. Agric. Food Chem. 2001, 49, 3622–3627. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jiang, L.; Rao, J.; Liu, Y.; Yang, L.; Zhang, D. Evaluation of Four Genes in Rice for Their Suitability As Endogenous Reference Standards in Quantitative PCR. J. Agric. Food Chem. 2010, 58, 11543–11547. [Google Scholar] [CrossRef]
- Papazova, N.; Zhang, D.; Gruden, K.; Vojvoda, J.; Yang, L.; Gašparič, M.B.; Blejec, A.; Fouilloux, S.; De Loose, M.; Taverniers, I. Evaluation of the reliability of maize reference assays for GMO quantification. Anal. Bioanal. Chem. 2010, 396, 2189–2201. [Google Scholar] [CrossRef]
- Broothaerts, W.; Corbisier, P.; Schimmel, H.; Trapmann, S.; Vincent, S.; Emons, H. A Single Nucleotide Polymorphism (SNP839) in the adh1 Reference Gene Affects the Quantitation of Genetically Modified Maize (Zea mays L.). J. Agric. Food Chem. 2008, 56, 8825–8831. [Google Scholar] [CrossRef] [PubMed]
- Bru, D.; Martin-Laurent, F.; Philippot, L. Quantification of the Detrimental Effect of a Single Primer-Template Mismatch by Real-Time PCR Using the 16S rRNA Gene as an Example. Appl. Environ. Microbiol. 2008, 74, 1660–1663. [Google Scholar] [CrossRef]
- Ghedira, R.; Papazova, N.; Vuylsteke, M.; Ruttink, T.; Taverniers, I.; De Loose, M. Assessment of Primer/Template Mismatch Effects on Real-Time PCR Amplification of Target Taxa for GMO Quantification. J. Agric. Food Chem. 2009, 57, 9370–9377. [Google Scholar] [CrossRef]
- Bhattramakki, D.; Dolan, M.; Hanafey, M.; Wineland, R.; Vaske, D.; Iii, J.C.R.; Tingey, S.V.; Rafalski, A. Insertion-deletion polymorphisms in 3′ regions of maize genes occur frequently and can be used as highly informative genetic markers. Plant Mol. Biol. 2002, 48, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Tenaillon, M.I.; Sawkins, M.C.; Long, A.D.; Gaut, R.L.; Doebley, J.F.; Gaut, B.S. Patterns of DNA sequence polymorphism along chromosome 1 of maize (Zea mays spp. mays L.). Proc. Natl. Acad. Sci. USA 2001, 98, 9161–9166. [Google Scholar] [CrossRef]
- Ching, A.; Caldwell, K.S.; Jung, M.; Dolan, M.; Smith, O.S.; Tingey, S.; Morgante, M.; Rafalski, A.J. SNP frequency, haplotype structure and linkage disequilibrium in elite maize inbred lines. BMC Genet. 2002, 3, 1–14. [Google Scholar] [CrossRef]
- Brunner, S.; Fengler, K.; Morgante, M.; Tingey, S.; Rafalski, A. Evolution of DNA sequence non-homologies among maize in-breds. Plant. Cell 2005, 17, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Bi, I.V.; McMullen, M.D.; Sanchez-Villeda, H.; Schroeder, S.; Gardiner, J.; Polacco, M.; Soderlund, C.; Wing, R.; Fang, Z.; Coe, E.H. Single Nucleotide Polymorphisms and Insertion–Deletions for Genetic Markers and Anchoring the Maize Fingerprint Contig Physical Map. Crop. Sci. 2006, 46, 12–21. [Google Scholar] [CrossRef]
- Buckler, E.S.; Gaut, B.S.; McMullen, M.D. Molecular and functional diversity of maize. Curr. Opin. Plant Biol. 2006, 9, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zhao, H.; Ren, L.; Song, W.; Zeng, B.; Guo, J.; Wang, B.; Liu, Z.; Chen, J.; Li, W.; et al. Genome-wide genetic changes during modern breeding of maize. Nat. Genet. 2012, 44, 812–815. [Google Scholar] [CrossRef]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The Effect of Primer-Template Mismatches on the Detection and Quantification of Nucleic Acids Using the 5′ Nuclease Assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Klungthong, C.; Chinnawirotpisan, P.; Hussem, K.; Phonpakobsin, T.; Manasatienkij, W.; Ajariyakhajorn, C.; Rungro-jcharoenkit, K.; Gibbons, R.V.; Jarman, R.G. The impact of primer and probe-template mismatches on the sensitivity of pan-demic influenza A/H1N1/2009 virus detection by real-time RT-PCR. J. Clin. Virol. 2010, 48, 91–95. [Google Scholar] [CrossRef]
- De Preter, K.; Speleman, F.; Combaret, V.; Lunec, J.; Laureys, G.; Eussen, B.H.; Francotte, N.; Board, J.; Pearson, A.D.; De Paepe, A.; et al. Quantification of MYCN, DDX1, and NAG Gene Copy Number in Neuroblastoma Using a Real-Time Quantitative PCR Assay. Mod. Pathol. 2002, 15, 159–166. [Google Scholar] [CrossRef]
- Wan, J.; Song, L.; Wu, Y.; Brzoska, P.; Keys, D.; Chen, C.; Valliyodan, B.; Shannon, J.G.; Nguyen, H.T. Application of Digital PCR in the Analysis of Transgenic Soybean Plants. Adv. Biosci. Biotechnol. 2016, 7, 403–417. [Google Scholar] [CrossRef]
- Huggett, J.F.; Novak, T.; Garson, J.; Green, C.; Morris-Jones, S.D.; Miller, R.F.; Zumla, A. Differential susceptibility of PCR reactions to inhibitors: An important and unrecognised phenomenon. BMC Res. 2008, 1, 70. [Google Scholar] [CrossRef]
- Taverniers, I.; Van Bockstaele, E.; De Loose, M. Cloned plasmid DNA fragments as calibrators for controlling GMOs: Different real-time duplex quantitative PCR methods. Anal. Bioanal. Chem. 2003, 378, 1198–1207. [Google Scholar] [CrossRef]
- Corbisier, P.; Broeders, S.; Charels, D.; Gioria, S.; Trapmann, S.; Vincent, S.; Emons, H. The Certification of Plasmidic DNA Containing MON 810 Maize DNA Fragments, Certified Reference Material ERM-AD413. EUR 22948 EN; European Communities: Luxembourg, 2007. [Google Scholar]
- Huang, H.-Y.; Pan, T.-M. Detection of Genetically Modified Maize MON810 and NK603 by Multiplex and Real-Time Polymerase Chain Reaction Methods. J. Agric. Food Chem. 2004, 52, 3264–3268. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guo, J.; Pan, A.; Zhang, H.; Zhang, K.; Wang, Z.; Zhang, D. Event-Specific Quantitative Detection of Nine Genetically Modified Maizes Using One Novel Standard Reference Molecule. J. Agric. Food Chem. 2006, 55, 15–24. [Google Scholar] [CrossRef]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Songkumarn, P.; Liu, J.; Wang, G.-L. A Versatile Zero Background T-Vector System for Gene Cloning and Functional Genomics. Plant Physiol. 2009, 150, 1111–1121. [Google Scholar] [CrossRef]
- Yue, Y.F.; Wu, G.; Wu, Y.H.; Lu, C.M. Development of qualitative PCR method targeting marker genes in transgenic plants. Chin. J. Oil Crop. Sci. 2011, 33, 280–289. [Google Scholar]
- Hernández, M.; Duplan, M.-N.; Berthier, G.; Vaïtilingom, M.; Hauser, W.; Freyer, R.; Pla, M.; Bertheau, Y. Development and Comparison of Four Real-Time Polymerase Chain Reaction Systems for Specific Detection and Quantification of Zea mays L. J. Agric. Food Chem. 2004, 52, 4632–4637. [Google Scholar] [CrossRef]
- Ginzinger, D.G. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 2002, 30, 503–512. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primers and Probe | Maize Cultivar | Linearity (R2) | PCR Efficiency (E) | Standard Curve |
|---|---|---|---|---|
| hmg-taq-F1/R1, hmg-taq-Probe | PH207 (T > A) | 0.997 | 1.030 | y = −3.251x + 33.984 |
| F7 (G > T) | 0.998 | 1.056 | y = −3.196x + 33.781 | |
| ND101 (without SNPs) | 0.995 | 0.995 | y = −3.336x + 33.105 | |
| hmg-taq-F2/R2, hmg-taq-Probe | PH207 (T > A) | 0.997 | 0.989 | y = −3.350x + 33.634 |
| F7 (G > T) | 0.997 | 1.001 | y = −3.319x + 33.451 | |
| ND101 (without SNPs) | 0.996 | 1.004 | y = −3.312x + 32.605 |
| Gene Name | Predicted Copy Number (Copies/μL) | Mean Ct Value | SD | CV (%) |
|---|---|---|---|---|
| bar gene | 107 | 16.13 | 0.48 | 2.94 |
| 106 | 19.47 | 0.46 | 2.35 | |
| 105 | 22.81 | 0.44 | 1.94 | |
| 104 | 26.23 | 0.45 | 1.71 | |
| 103 | 29.64 | 0.52 | 1.18 | |
| hmg gene | 107 | 15.02 | 0.28 | 1.88 |
| 106 | 18.35 | 0.31 | 1.68 | |
| 105 | 21.69 | 0.30 | 1.36 | |
| 104 | 25.11 | 0.30 | 1.19 | |
| 103 | 28.44 | 0.40 | 1.40 |
| Copy Number | Number of Samples Determined by TaqMan (% of total) | Correlation with Southern Blot | Correlation with dPCR | ||
|---|---|---|---|---|---|
| Number of Assayed Samples | Number of Consistent Samples (%) | Number of Assayed Samples | Number of Consistent Samples (%) | ||
| 0 | 2 (4.4%) | 2 | 2 (100.0%) | ||
| 1 | 33 (73.3%) | 33 | 30 (90.9%) | 5 | 5 (100.0%) |
| 2 | 6 (13.3%) | 6 | 4 (66.7%) | 2 | 2 (100.0%) |
| >2 | 4 (9.0%) | 4 | 1 (25.0%) | 1 | 1 (100.0%) |
| Total | 45 | 45 | 8 | ||
| Copy Number | Accuracy of Technical Repetitions | ||
|---|---|---|---|
| r = 1 | r = 2 | r = 3 | |
| 1 | 0.9673 | 0.9820 | 1.0000 (assumed) |
| 2 | 0.8932 | 0.9431 | 1.0000 (assumed) |
| Copy Number | Number of T0 Samples Determined by TaqMan Assay |
Real Copy Number of T0 Samples Verified by T1 and T2 Progeny |
|---|---|---|
| Number of Consistent Samples (%) | ||
| 1 | 2016 | 1931 (95.8%) |
| 2 | 74 | |
| >2 | 11 | |
| Total | 2016 | 2016 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, F.; Cheng, J.; Liu, X.; Wang, X.-Q. High-Throughput and Accurate Determination of Transgene Copy Number and Zygosity in Transgenic Maize: From DNA Extraction to Data Analysis. Int. J. Mol. Sci. 2021, 22, 12487. https://doi.org/10.3390/ijms222212487
Liu F, Cheng J, Liu X, Wang X-Q. High-Throughput and Accurate Determination of Transgene Copy Number and Zygosity in Transgenic Maize: From DNA Extraction to Data Analysis. International Journal of Molecular Sciences. 2021; 22(22):12487. https://doi.org/10.3390/ijms222212487
Chicago/Turabian StyleLiu, Fang, Jinkui Cheng, Xuhua Liu, and Xi-Qing Wang. 2021. "High-Throughput and Accurate Determination of Transgene Copy Number and Zygosity in Transgenic Maize: From DNA Extraction to Data Analysis" International Journal of Molecular Sciences 22, no. 22: 12487. https://doi.org/10.3390/ijms222212487
APA StyleLiu, F., Cheng, J., Liu, X., & Wang, X.-Q. (2021). High-Throughput and Accurate Determination of Transgene Copy Number and Zygosity in Transgenic Maize: From DNA Extraction to Data Analysis. International Journal of Molecular Sciences, 22(22), 12487. https://doi.org/10.3390/ijms222212487