
Identification of Co-Expressed Genes Related to Theacrine Synthesis in Tea Flowers at Different Developmental Stages

 and
and
Abstract
:1. Introduction
2. Results
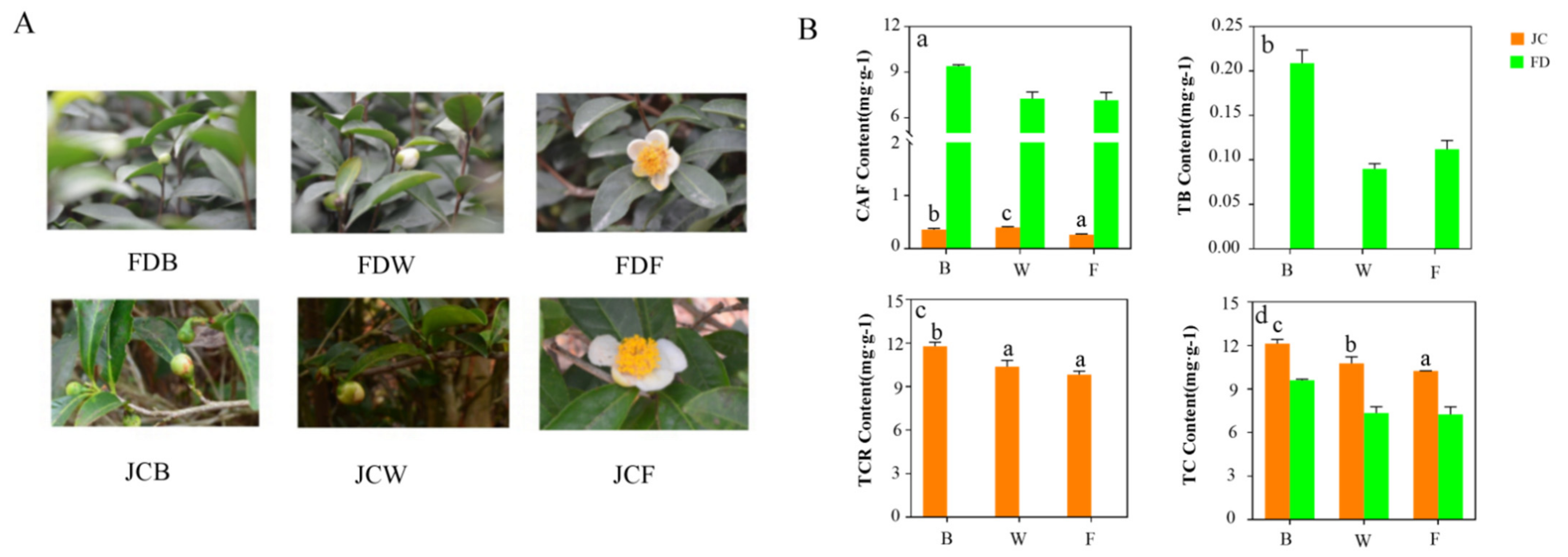
2.1. Quantification of Purine Alkaloids in Different Flowering Periods between JC and FD
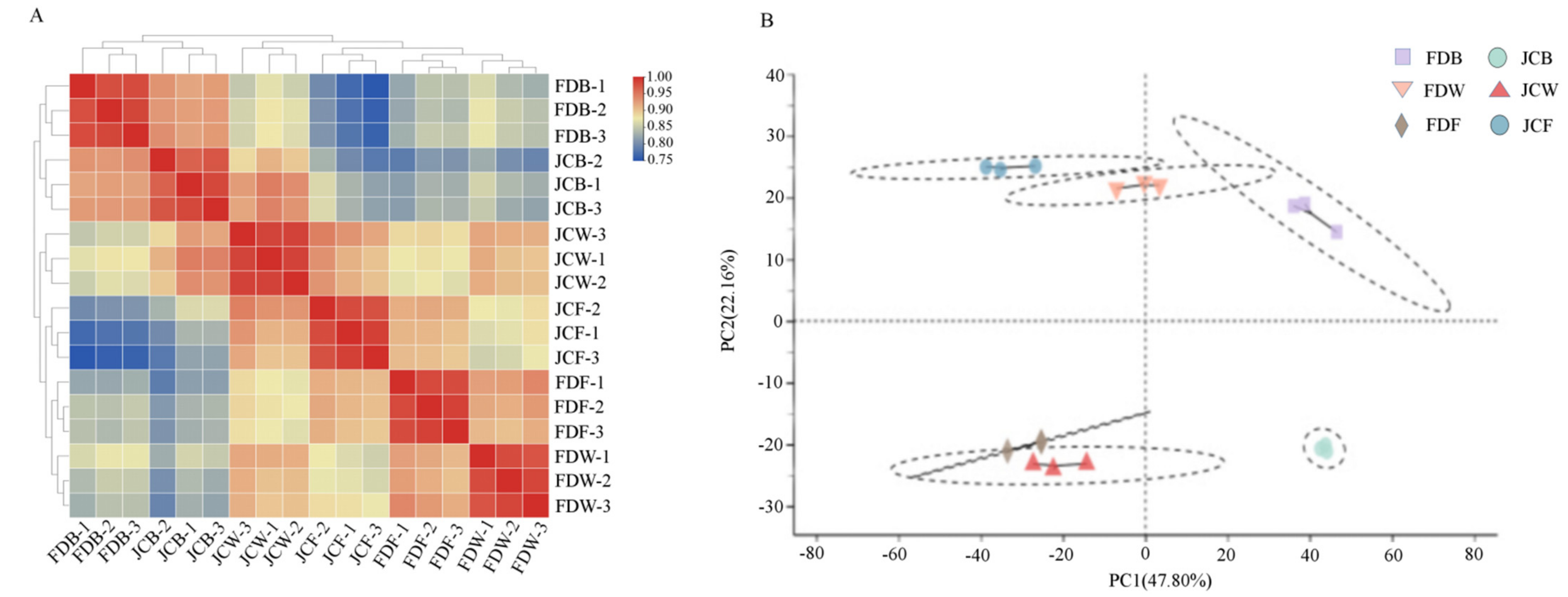
2.2. Transcriptome Data Analysis
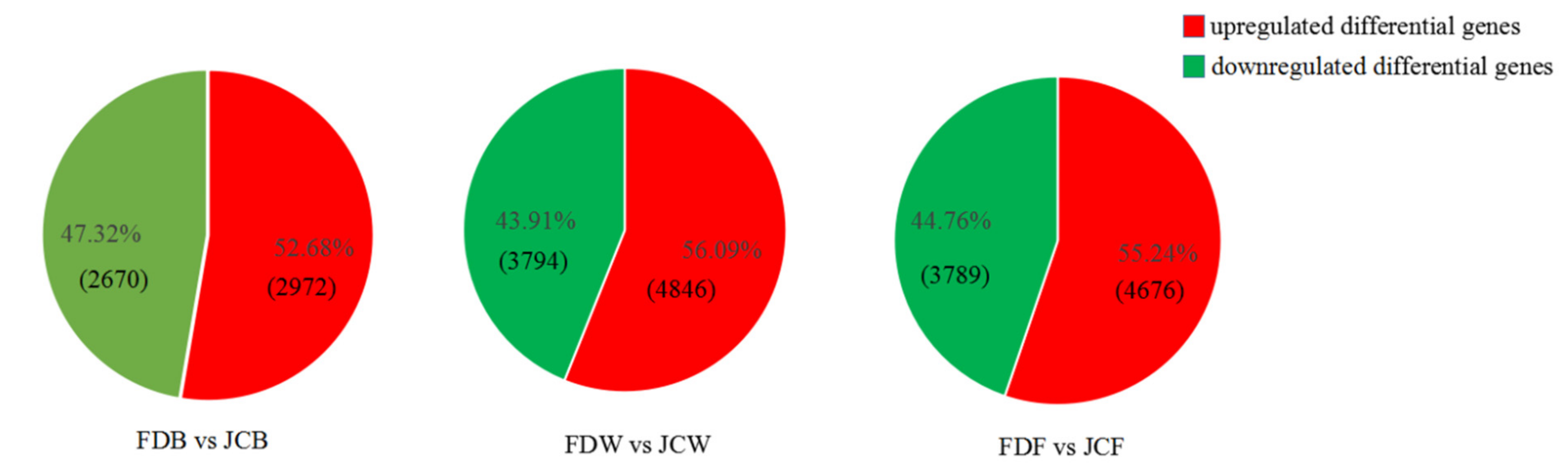
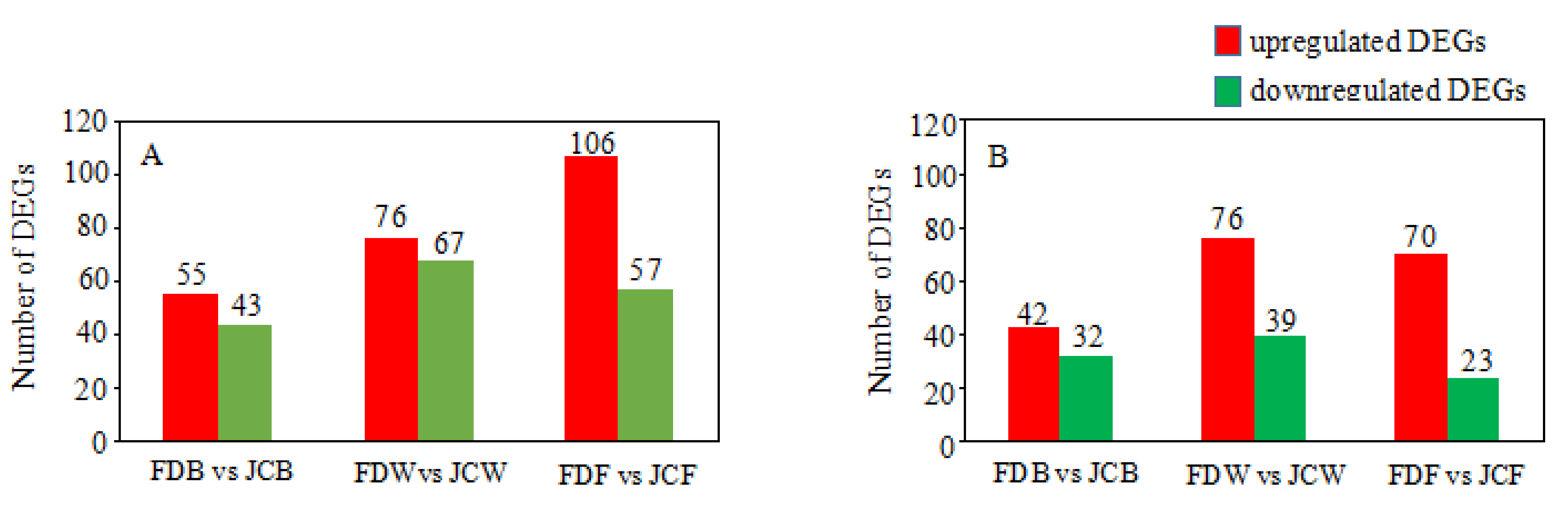
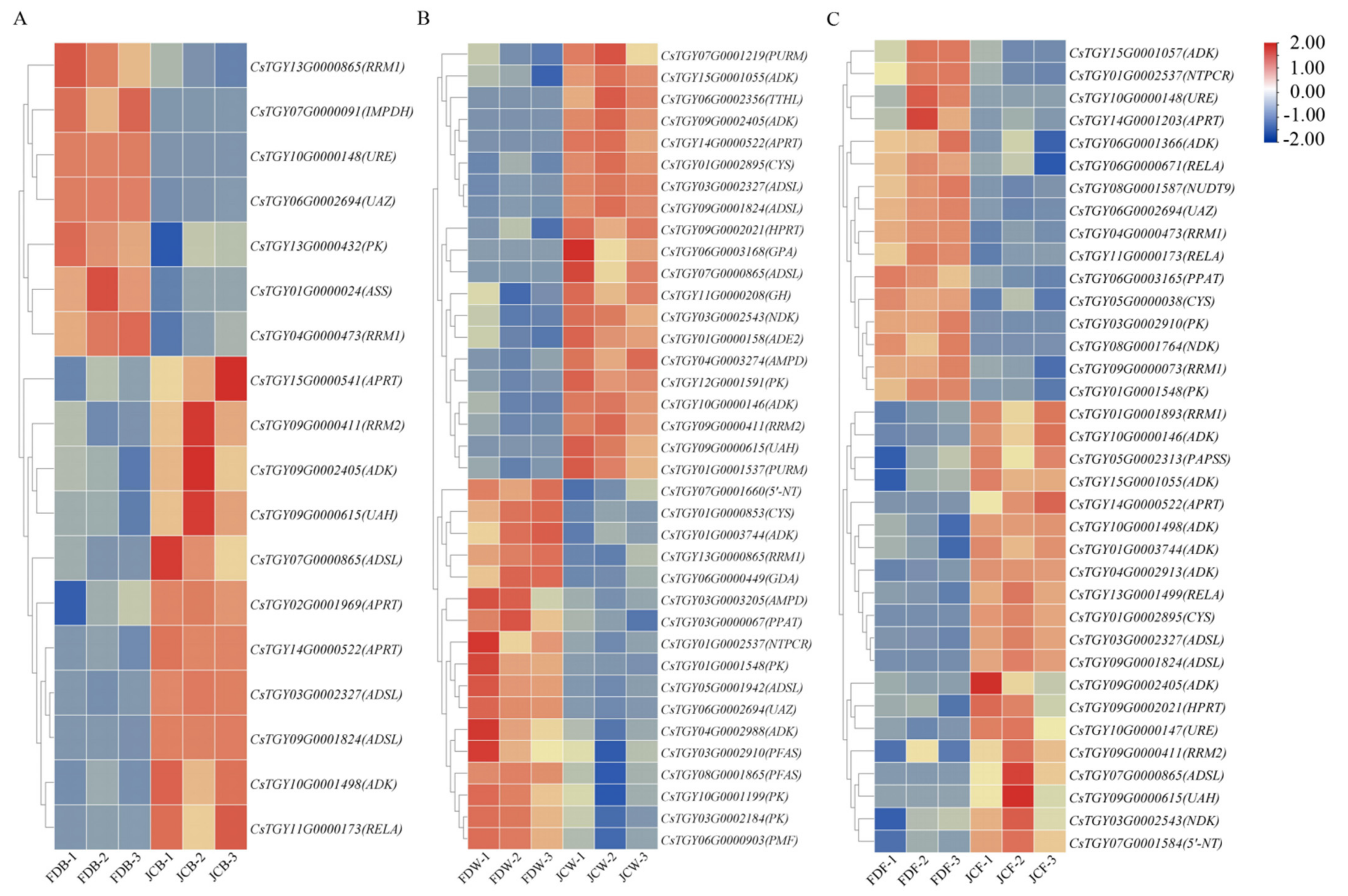
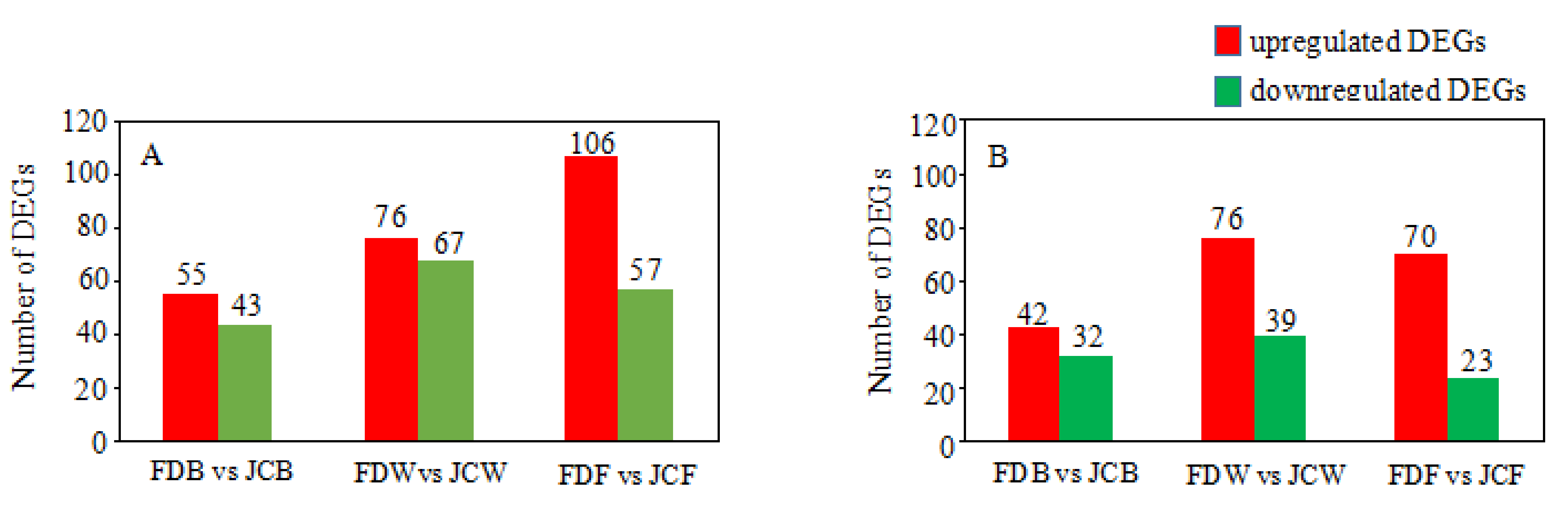
2.3. Identification of Differentially Expressed Genes (DEGs)
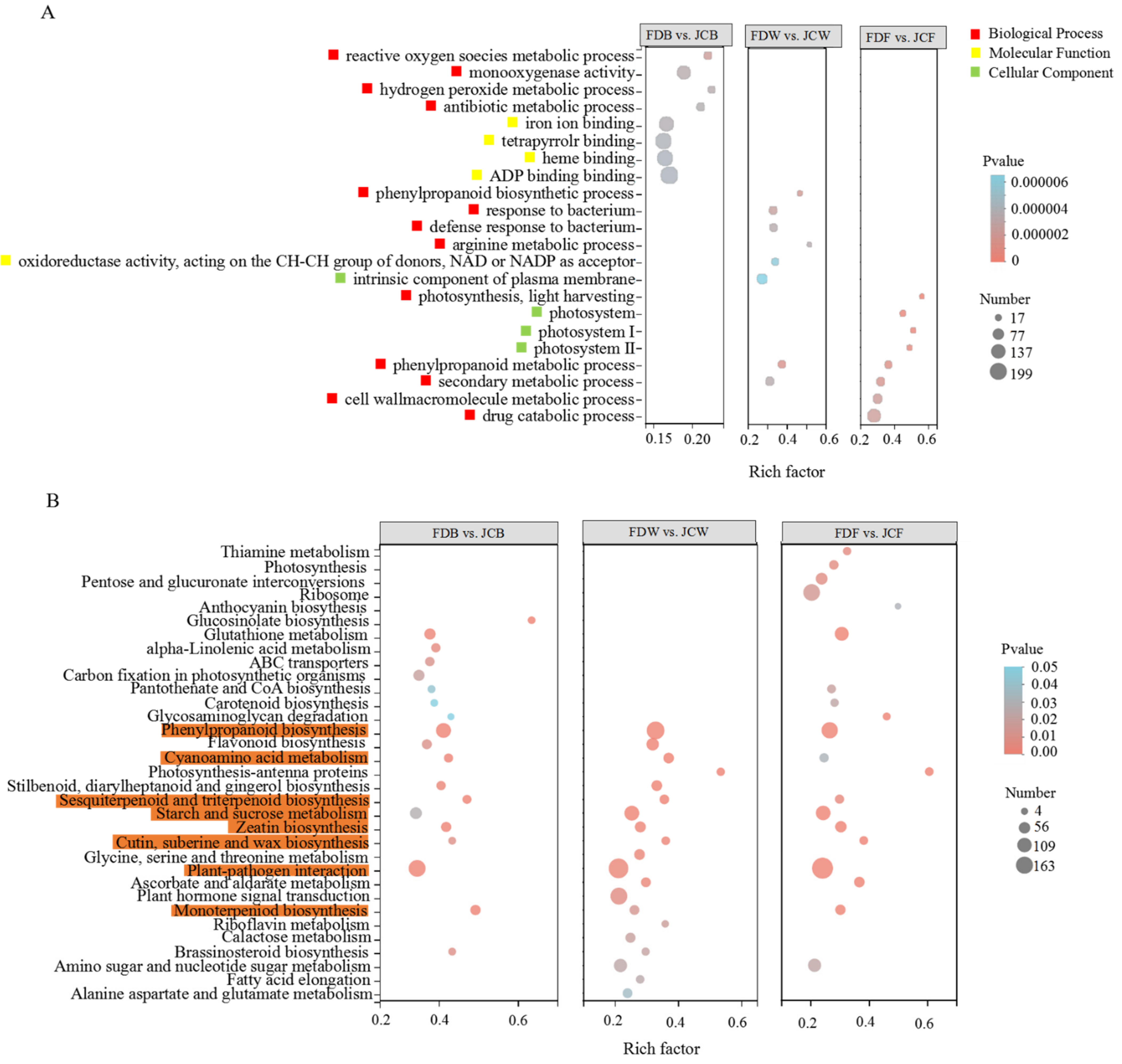
2.4. GO and KEGG Enrichment Analysis of DEGs
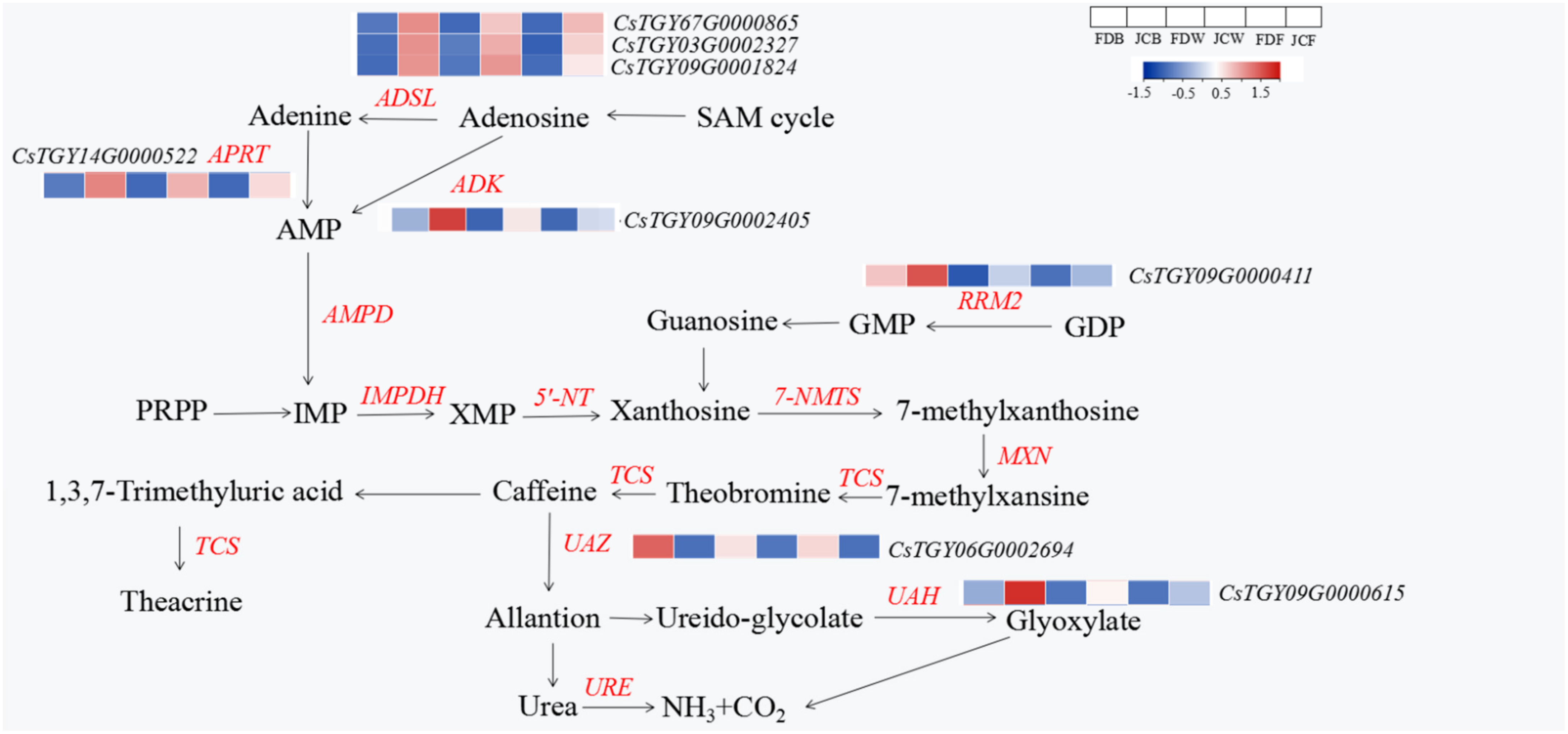
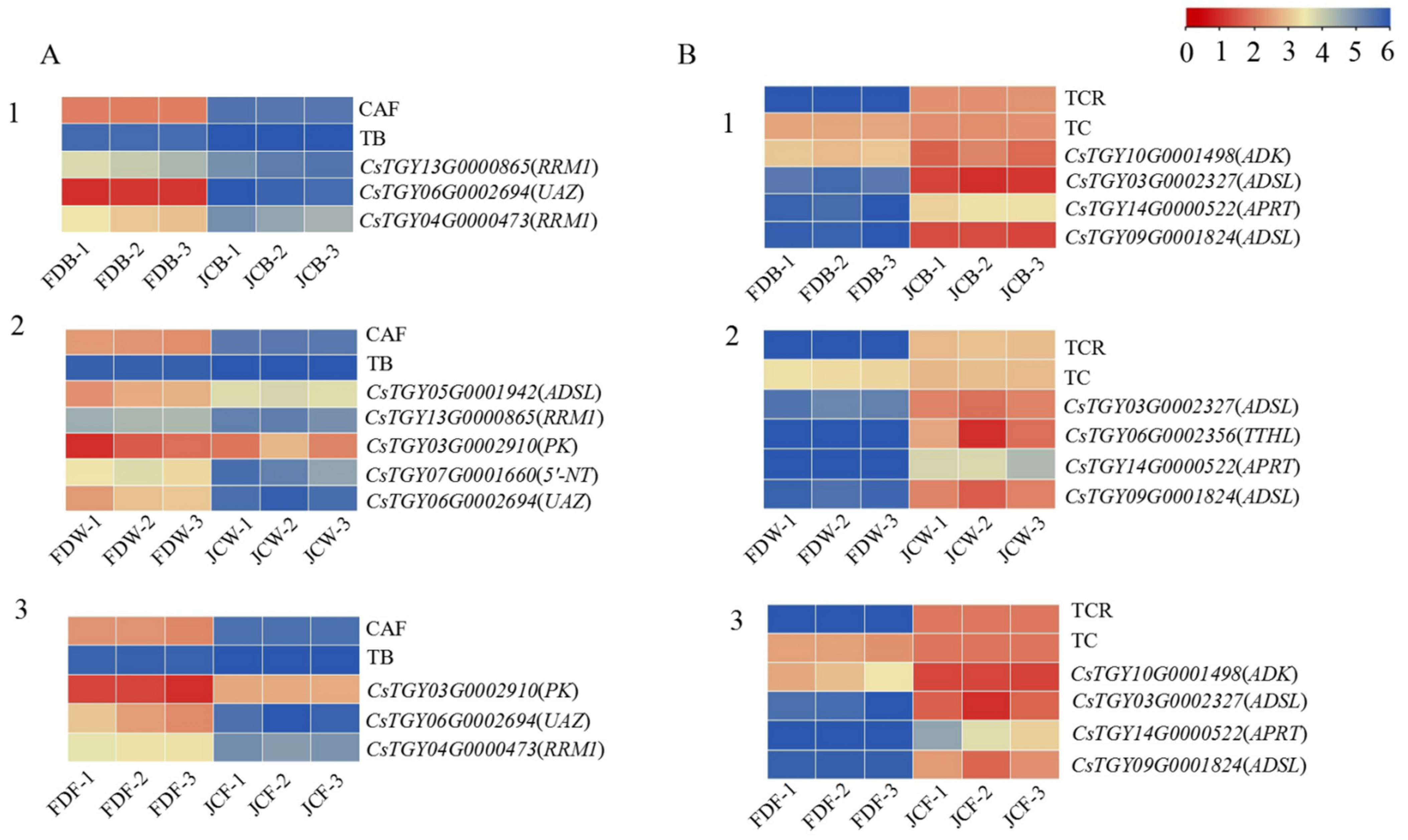
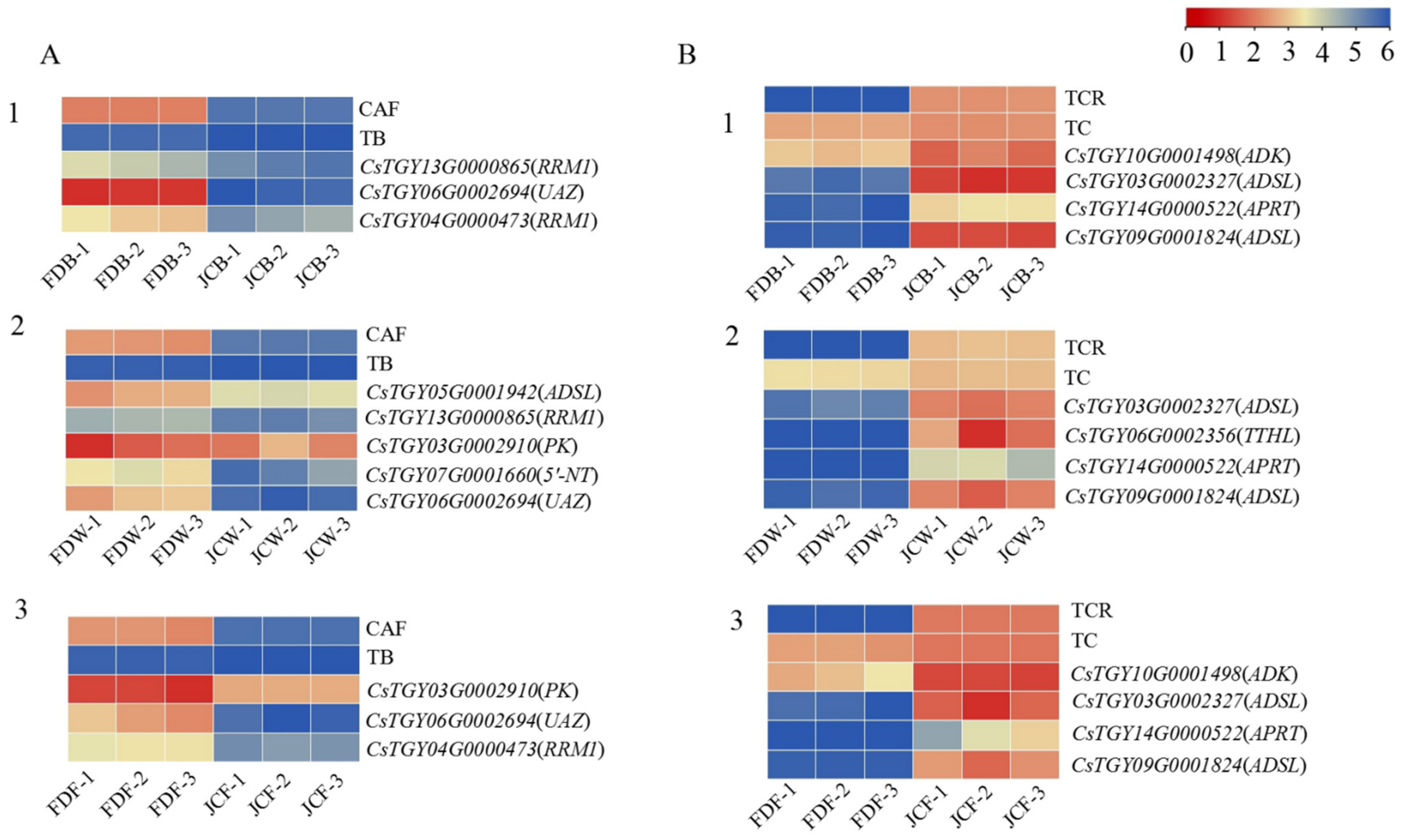
2.5. DEGs Analysis in the Purine Alkaloid Pathway
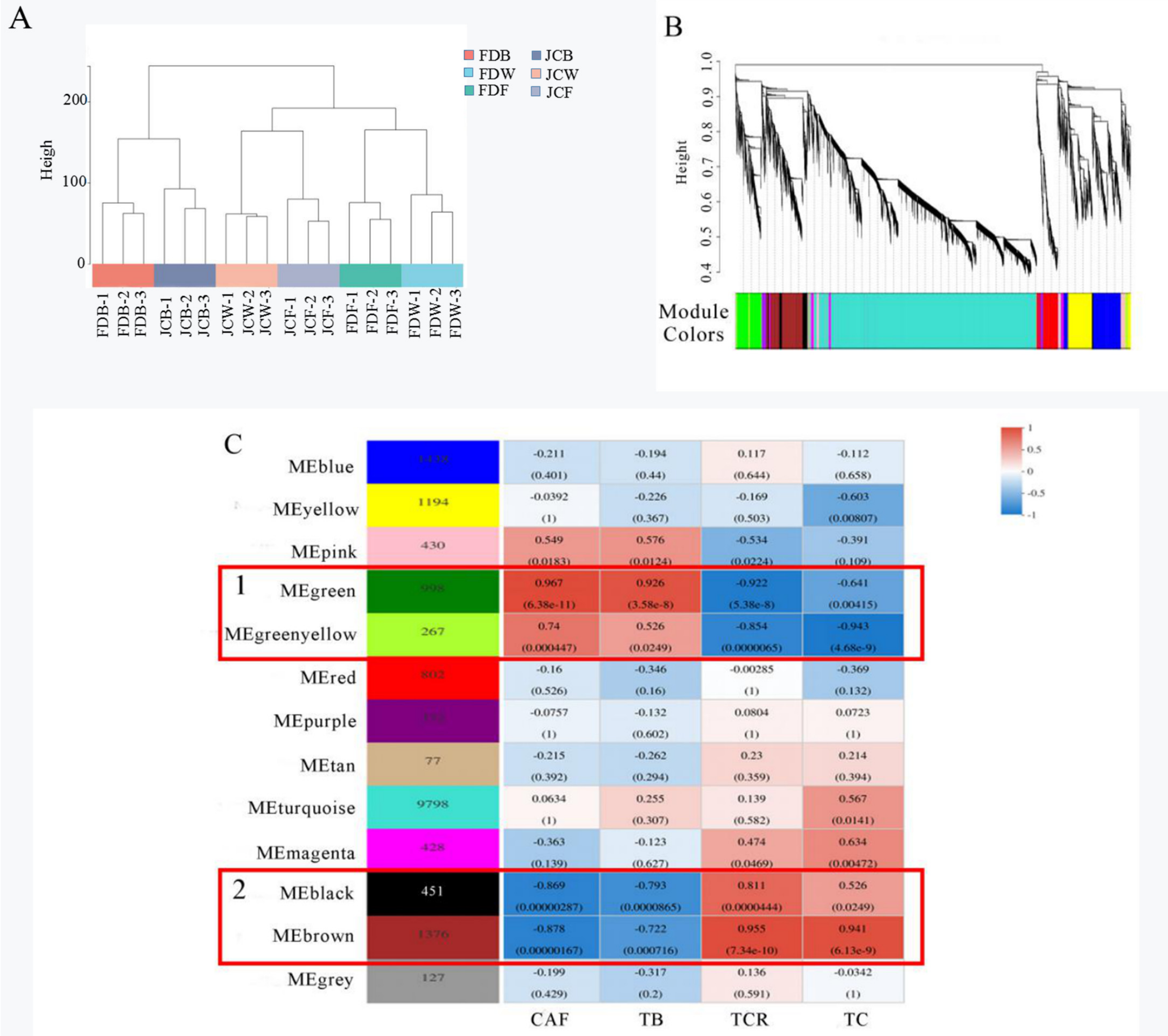
2.6. Co-Expression Network Analysis
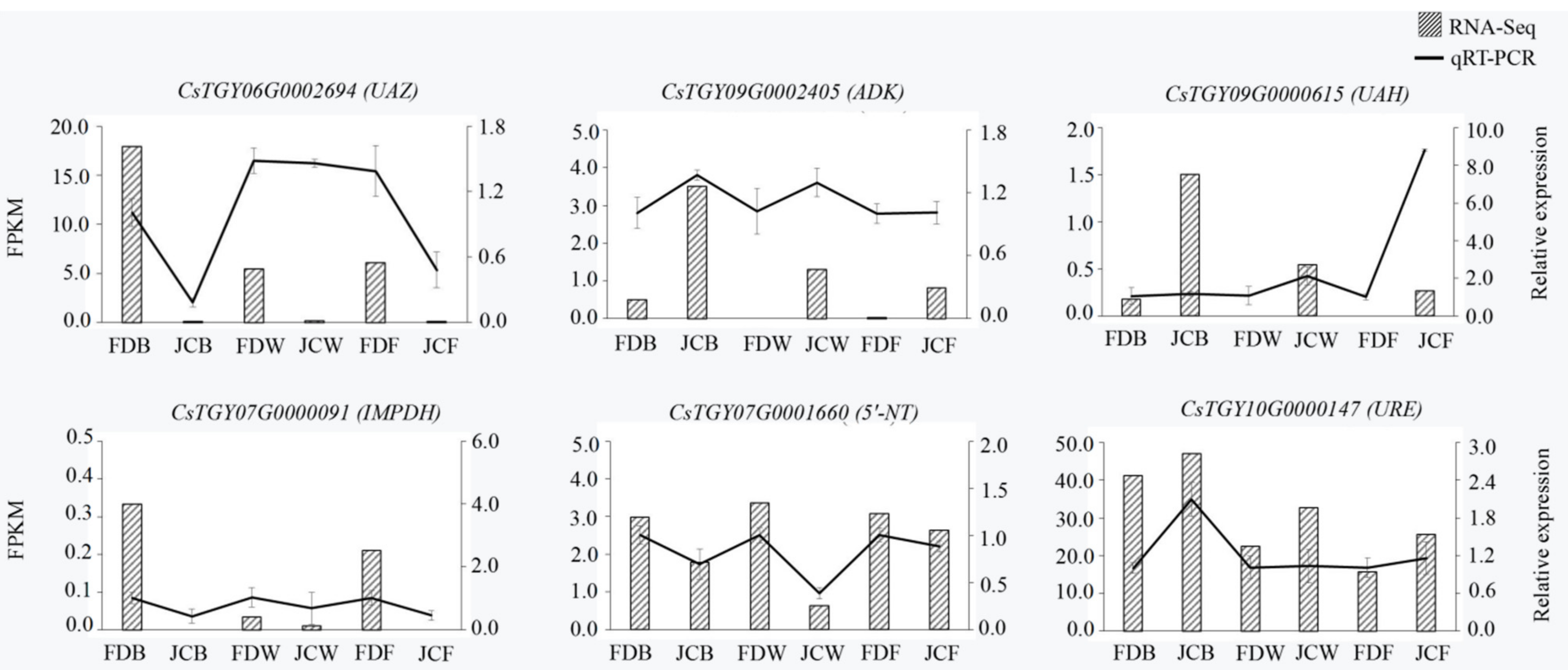
2.7. Analysis of DEG Expression Level by PCR
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Determination of Purine Alkaloids
4.3. Acquisition and Analysis of Transcriptome Sequencing
4.4. Differentially Expressed Genes and Enrichment Analysis
4.5. Coexpression Module Construction and Analysis
4.6. Verification of Transcriptome Data
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Li, H.J.; Fang, K.X.; Qin, D.D.; Jiang, X.H.; Wang, Q.S.; Pan, C.D.; Li, B.; Wang, Q.; Wu, H.L. Comparative Transcriptome Analysis Reveals Putative Genes Responsible for High Theacrine Content in Kucha (Camellia kucha (Chang et Wang) Chang). Trop. Plant Biol. 2021, 14, 1–11. [Google Scholar] [CrossRef]
- Dai, L.F.; Luo, L.Y.; Luo, J.Q.; Chan, R.; Liu, Y.; Zeng, L. Overview of Bitter Substances in Plants and Their Application in Food Industry. J. Chin. Inst. Food Sci Tech. 2020, 20, 305–318. [Google Scholar]
- Ouyang, S.-H.; Zhai, Y.-J.; Wu, Y.-P.; Xie, G.; Wang, G.-E.; Mao, Z.-F.; Hu, H.-H.; Luo, X.-H.; Sun, W.-Y.; Liang, L.; et al. Theacrine, a Potent Antidepressant Purine Alkaloid from a Special Chinese Tea, Promotes Adult Hippocampal Neurogenesis in Stressed Mice. J. Agric. Food Chem. 2021, 69, 7016–7027. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.X.; Lin, Y.C.; Su, J.Y.; Song, X.H.; Zhang, H.D. The purine alkaloid of Camellia assamica var kucha Chang et Wang. Zhongshan Uni. Nat. Sci. 1999, 5, 82–86. [Google Scholar]
- Zheng, X.Q.; Ye, C.X.; Misako, K.; Alan, C.; Hiroshi, A. Theacrine (1,3,7,9-tetramethyluric acid) synthesis in leaves of a Chinese tea, kucha (Camellia assamica var. kucha). Phytochemistry 2002, 60, 129–134. [Google Scholar] [CrossRef]
- Yang, P.D.; Liu, Z.; Zhao, Y.; Cheng, Y.; Ning, J.; Liu, Y.; Huang, J.A.; Yang, Y. Strain evaluation of Jianghua bitter tea based on agronomic traits and SSR marker relationship analysis. Plant. Breed. 2021, 19, 2402–2409. [Google Scholar]
- Li, Y.-F.; Ouyang, S.-H.; Chang, Y.-Q.; Wang, T.-M.; Li, W.-X.; Tian, H.-Y.; Cao, H.; Kurihara, H.; He, R.-R. A comparative analysis of chemical compositions in Camellia sinensis var. puanensis Kurihara, a novel Chinese tea, by HPLC and UFLC-Q-TOF-MS/MS. Food Chem. 2017, 216, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.D.; Wang, Q.S.; Li, H.J.; Jiang, X.H.; Fang, K.X.; Wang, Q.; Li, B.; Pan, C.D.; Wu, H.L. Identification of key metabolites based on non-targeted metabolomics and chemometrics analyses provides insights into bitterness in Kucha [Camellia kucha (Chang et Wang) Chang]. Food Res. Int. 2020, 138, 109789. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Qin, D.D.; Jiang, X.H.; Fang, K.X.; Wang, Q.S.; Pan, C.D.; Li, B.; Wang, Q.; Wu, H.L. Analysis and evaluation of purine alkaloid contents in bitter tea germplasm resources from Guangdong. Tea sci. 2021, 41, 337–349. [Google Scholar]
- Pang, D.D.; Liu, Y.F.; Sun, Y.N.; Tian, Y.P.; Song, W.X.; Chen, L.B. Full-Length transcriptome and gene structure analysis of Kucha (Camellia sinensis). Acta Agric. Boreali-Occident. Sin. 2021, 30, 563–571. [Google Scholar]
- Qin, D.D.; Wang, Q.S.; Li, H.J.; Fang, K.X.; Jiang, X.H.; Pan, C.D.; Wang, Q.; Li, B.; Wu, H.L. Research progresses on Camellia kucha (Chang et Wang) Chang and theacrine. Food sci. 2021, 42, 353–359. [Google Scholar]
- Wang, S.L.; Chen, J.D.; Ma, J.Q.; Jin, J.Q.; Chen, L.; Yao, M.Z. Novel insight into theacrine metabolism revealed by tran-scriptome analysis in bitter tea (Kucha, Camellia sinensis). Sci. Rep. 2020, 10, 6286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Li, Y.-F.; Wang, Y.J.; Tan, L.; Cao, Z.-Q.; Xie, C.; Xie, G.; Gong, H.-B.; Sun, W.-Y.; Ouyang, S.-H.; et al. Identification and characterization of N9-methyltransferase involved in converting caffeine into non-Stimulatory theacrine in tea. Nat. Commun. 2020, 11, 1473. [Google Scholar] [CrossRef]
- Chen, X.M.; Wang, P.J.; Wang, S.Y.; Yang, R.X.; Sun, J.; Guo, Y.C.; Chen, X.J.; Zhao, F.; Ye, N.X. Excavation of genes involved in theacrine biosynthesis of Jiaochengkucha based on transcriptome. Chin. J. Appl. Envir Bio. 2021, 27, 1382–1389. [Google Scholar]
- Chen, D.; Chen, G.J.; Sun, Y.; Zeng, X.X.; Ye, H. Physiological genetics, chemical composition, health benefits and toxicology of tea (Camellia sinensis L.) flower: A review. Food Res. Int. 2020, 137, 109584. [Google Scholar] [CrossRef]
- Han, Q.; Xiong, C.Y.; Shi, J.; Gao, Y.; Chen, Y.S.; Ling, Z.J.; He, P.M. Isolation, chemical characterization and antioxidant ac-tivities of a water-solublepolysaccharide fraction of tea (Camelllia sinensis) flower. J. Food Biochem. 2012, 36, 46–55. [Google Scholar] [CrossRef]
- Hamao, M.; Matsuda, H.; Nakamura, S.; Nakashima, S.; Semura, S.; Maekubo, S.; Yoshikawa, M. Anti-Obesity effects of the methanolic extract and chakasa-ponins from the flower buds of Camellia sinensis in mice. Med. Chem. 2011, 19, 6033–6041. [Google Scholar]
- Zhao, X.; Gu, Y.P.; Qian, H. Development of Ice Tea Flower Drink. J. Anhui. Agric. Sci. 2010, 7, 2924–2925. [Google Scholar]
- Zhang, W.J.; Lin, C.; Lin, S.; Ji, A.B.; Zheng, T.T.; Ma, L.; Peng, W.S.; Gong, W.Y.; Yan, L. Making technology of tea tree flower soft candy. China Food Saf. Magazine. 2016, 27, 146–148. [Google Scholar]
- Wei, X.L.; Chen, M.A.; Xiao, J.B.; Liu, Y.; Yu, L.; Zhang, H.; Wang, Y.F. Composition and bioactivity of tea flower polysac-charides obtained by differentmethods. Carbohydr. Polym. 2010, 79, 418–422. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Zhou, Y.; Zeng, L.T.; Dong, F.; Tu, Y.Y.; Yang, Z.Y. Occurrence of functional molecules in the flowers of tea (Camellia sinensis) plants: Evidence for asecond resource. Molecules 2018, 23, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.T.; Wang, Y.; Ding, Z.T.; Liu, F. The dynamic changes of catechins and related genes in tea (Camellia sinensis) flowers. Acta Physiol. Plant. 2019, 41, 30. [Google Scholar] [CrossRef]
- Fan, K.; Zhang, Q.F.; Liu, M.Y.; Ma, L.F.; Shi, Y.Z.; Ruan, J.Y. Metabolomic and transcriptional analyses reveal the mechanism of C, N allocation from source leaf to flower in tea plant (Camellia sinensis L.). J. Plant Physiol. 2019, 232, 200–208. [Google Scholar] [CrossRef]
- Rothenberg, O.D.; Yang, H.; Chen, M.; Zhang, W.; Zhang, L. Metabolome and Transcriptome Sequencing Analysis Reveals Anthocyanin Metabolism in Pink Flowers of Anthocyanin-Rich Tea (Camellia sinensis). Molecules 2019, 24, 1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-S.; Wu, S.-S.; Lin, J.-K. Determination of Tea Polyphenols and Caffeine in Tea Flowers (Camellia sinensis) and Their Hydroxyl Radical Scavenging and Nitric Oxide Suppressing Effects. J. Agric. Food Chem. 2003, 51, 975–980. [Google Scholar] [CrossRef]
- Xia, E.H.; Zhang, H.B.; Sheng, J.; Li, K.; Zhang, Q.J.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant. 2017, 10, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.L.; Yang, H.; Wang, S.B.; Zhao, J.; Liu, C.; Gao, L.P.; Xia, E.H.; Lu, Y.; Tai, Y.L.; She, G.; et al. Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 2018, 115, E4151–E4158. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.J.; Yu, J.X.; Jin, S.; Chen, S.; Yue, C.; Wang, W.L.; Gao, S.L.; Cao, H.L.; Zheng, Y.C.; Gu, M.Y.; et al. Genetic basis of high aroma and stress tolerance in the oolong tea cultivar genome. Hortic. Res. 2021, 8, 107. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.Y.; Sun, C.; Bi, Z.Z.; Liang, W.J.; Li, P.C.; Zhang, J.L.; Bai, J.P. Identification of drought-related co-expression modules and hub genes in potato roots based on WGCNA. Acta Agron. Sin. 2020, 46, 1033–1051. [Google Scholar] [CrossRef]
- Wang, P.J.; Jin, S.; Chen, X.J.; Wu, L.Y.; Zheng, Y.C.; Yue, C.; Guo, Y.C.; Zhang, X.T.; Yang, J.F.; Ye, N.Y. Chromatin accessibility and translational landscapes of tea plants under chilling stress. Hortic. Res. 2021, 8, 96. [Google Scholar] [CrossRef]
- Zhang, X.T.; Chen, S.; Shi, L.Q.; Gong, D.P.; Zhang, S.C.; Zhao, Q.; Zhan, D.L.; Vasseur, L.; Wang, Y.B.; Yu, J.X.; et al. Haplotype-Resolved genome assembly pro-vides insights into evolutionary history of the tea plant Camellia sinensis. Nat. Genet. 2021, 53, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. BMC Bioinform. 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.H.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; A Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Correction: Corrigendum: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.J.; Chen, S.R.; Gu, M.Y.; Chen, X.M.; Chen, X.J.; Yang, J.F.; Zhao, F.; Ye, N.X. Exploration of the Effects of Different Blue LED Light Intensities on Flavonoid and Lipid Metabolism in Tea Plants via Transcriptomics and Metabolomics. Int. J. Mol. Sci. 2020, 21, 4606. [Google Scholar] [CrossRef]
- Wu, Z.J.; Tian, C.; Jiang, Q.; Li, X.H.; Zhuang, J. Selection of suitable reference genes for qRT-PCR normalization during leaf development and hormonal stimuli in tea plant (Camellia sinensis). Sci. Rep. 2016, 6, 19748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Raw Reads (Gb) | Clean Reads (Gb) | Clean Bases (Gb) | Q20 (%) | Q30 (%) | GC Content (%) | Uniquely Mapped (%) |
|---|---|---|---|---|---|---|---|
| FDB-1 | 53.31 | 52.96 | 7.61 | 98.51 | 95.27 | 45.14 | 85.54 |
| FDB-2 | 54.64 | 54.30 | 7.80 | 98.59 | 95.52 | 44.88 | 85.29 |
| FDB-3 | 51.87 | 51.54 | 7.42 | 98.52 | 95.32 | 45.07 | 84.83 |
| Average | 53.27 | 52.93 | 7.61 | 98.54 | 95.37 | 45.03 | 85.22 |
| FDW-1 | 59.03 | 58.66 | 8.49 | 98.5 | 95.26 | 45.3 | 86.27 |
| FDW-2 | 61.01 | 60.64 | 8.78 | 98.54 | 95.39 | 45.52 | 86.44 |
| FDW-3 | 58.60 | 58.22 | 8.37 | 98.53 | 95.39 | 45.75 | 85.76 |
| Average | 59.55 | 59.17 | 8.55 | 98.52 | 95.35 | 45.52 | 86.16 |
| FDF-1 | 55.09 | 54.79 | 7.91 | 98.53 | 95.37 | 45.34 | 85.15 |
| FDF-2 | 56.64 | 56.31 | 8.09 | 98.52 | 95.35 | 45.16 | 84.48 |
| FDF-3 | 54.08 | 53.74 | 7.72 | 98.47 | 95.21 | 45.57 | 83.30 |
| Average | 55.27 | 54.95 | 7.91 | 98.51 | 95.31 | 45.36 | 84.31 |
| JCB-1 | 53.94 | 53.50 | 7.63 | 98.61 | 95.61 | 44.53 | 83.06 |
| JCB-2 | 57.28 | 56.86 | 8.15 | 98.6 | 95.58 | 44.42 | 83.23 |
| JCB-3 | 56.09 | 55.65 | 7.96 | 98.6 | 95.57 | 44.54 | 83.12 |
| Average | 55.77 | 55.34 | 7.91 | 98.60 | 95.59 | 44.50 | 83.14 |
| JCW-1 | 54.16 | 53.77 | 7.69 | 98.63 | 95.65 | 44.75 | 83.48 |
| JCW-2 | 49.22 | 48.77 | 6.93 | 98.51 | 95.4 | 44.34 | 83.51 |
| JCW-3 | 62.65 | 62.15 | 8.83 | 98.54 | 95.43 | 44.97 | 83.97 |
| Average | 55.34 | 54.90 | 7.82 | 98.56 | 95.49 | 44.69 | 83.65 |
| JCF-1 | 57.85 | 57.46 | 8.33 | 98.49 | 95.3 | 45.78 | 79.05 |
| JCF-2 | 52.88 | 52.50 | 7.55 | 98.48 | 95.27 | 45.48 | 80.13 |
| JCF-3 | 57.21 | 56.81 | 8.16 | 98.45 | 95.19 | 45.93 | 78.60 |
| Average | 55.98 | 55.59 | 8.01 | 98.47 | 95.25 | 45.73 | 79.26 |
| Purine Alkaloid | Retention Time (min) | Precurser Ion (m/z) | Product Ion (m/z) | Decluster Voltage (e/V) | Collision Energy (e/V) |
|---|---|---|---|---|---|
| Caffeine (CAF) | 2.55 | 195.2 | 138.1 a/110.2/83.0 | 80.0 | 25.0/31.0/35.0 |
| Theacrine (TCR) | 2.47 | 225.1 | 168.1 a/153.2/210.1 | 80.0 | 27.2/34.0/27.3 |
| Theobromine (TB) | 1.85 | 181.0 | 107.9 a /122.1/67.10 | 91.3 | 30.0/35.0/43.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Shao, S.; Yang, R.; Gu, M.; Wang, P.; Zhao, F.; Ye, N. Identification of Co-Expressed Genes Related to Theacrine Synthesis in Tea Flowers at Different Developmental Stages. Int. J. Mol. Sci. 2021, 22, 13394. https://doi.org/10.3390/ijms222413394
Chen X, Shao S, Yang R, Gu M, Wang P, Zhao F, Ye N. Identification of Co-Expressed Genes Related to Theacrine Synthesis in Tea Flowers at Different Developmental Stages. International Journal of Molecular Sciences. 2021; 22(24):13394. https://doi.org/10.3390/ijms222413394
Chicago/Turabian StyleChen, Xiaomin, Shuxian Shao, Ruxing Yang, Mengya Gu, Pengjie Wang, Feng Zhao, and Naixing Ye. 2021. "Identification of Co-Expressed Genes Related to Theacrine Synthesis in Tea Flowers at Different Developmental Stages" International Journal of Molecular Sciences 22, no. 24: 13394. https://doi.org/10.3390/ijms222413394
APA StyleChen, X., Shao, S., Yang, R., Gu, M., Wang, P., Zhao, F., & Ye, N. (2021). Identification of Co-Expressed Genes Related to Theacrine Synthesis in Tea Flowers at Different Developmental Stages. International Journal of Molecular Sciences, 22(24), 13394. https://doi.org/10.3390/ijms222413394