

Detection of a Novel Gull-like Clade of Newcastle Disease Virus and H3N8 Avian Influenza Virus in the Arctic Region of Russia (Taimyr Peninsula)
 , , , , , , , ,
, , , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
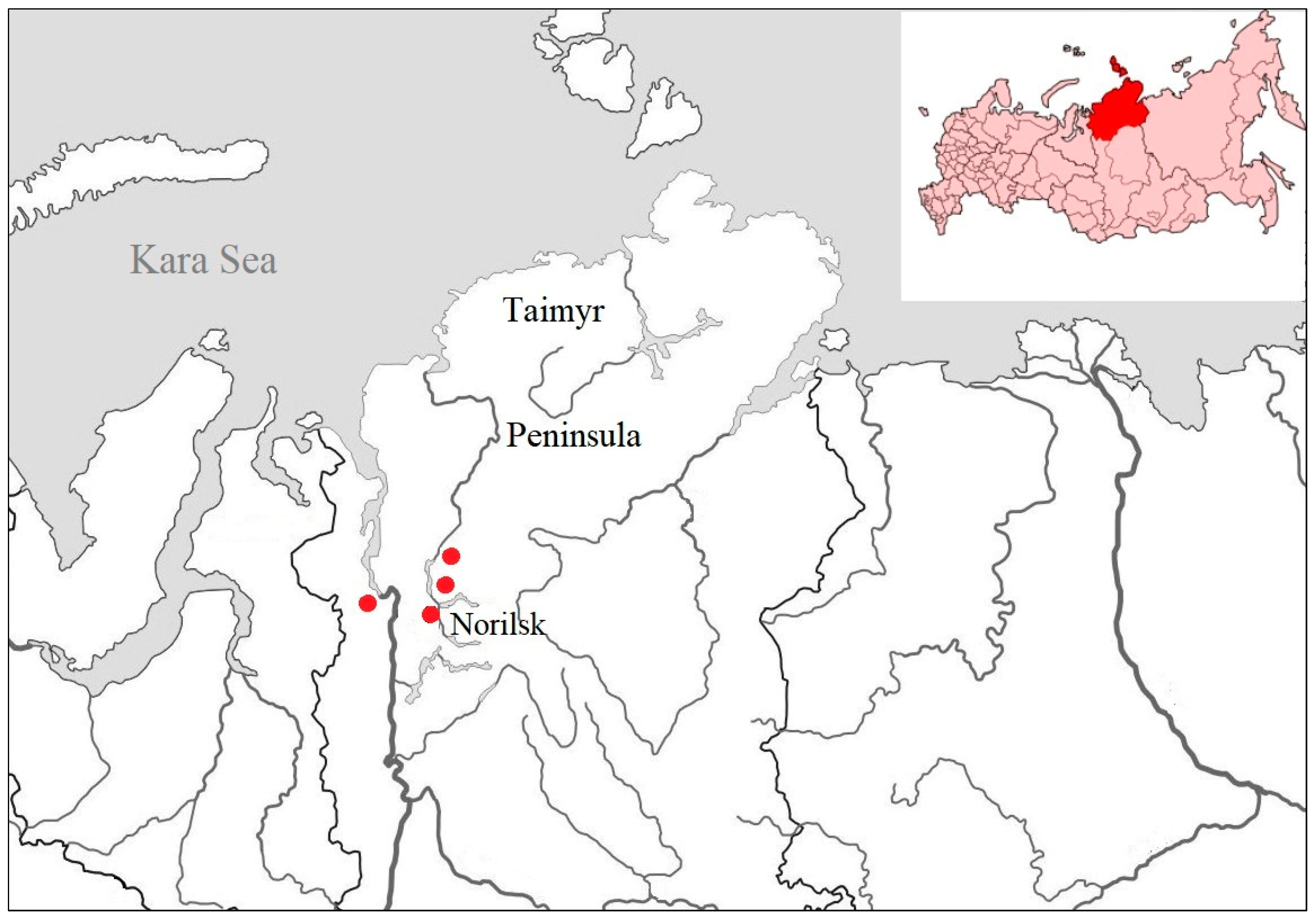
2.1. Sampling
2.2. Virus Isolation
2.3. Avian Paramyxovirus and Avian Influenza Virus Detection
2.4. Sequencing
2.5. Phylogenetic Analysis
2.5.1. Newcastle Disease Virus
2.5.2. Avian Influenza Virus
3. Results
3.1. Sampling and Virus Isolation
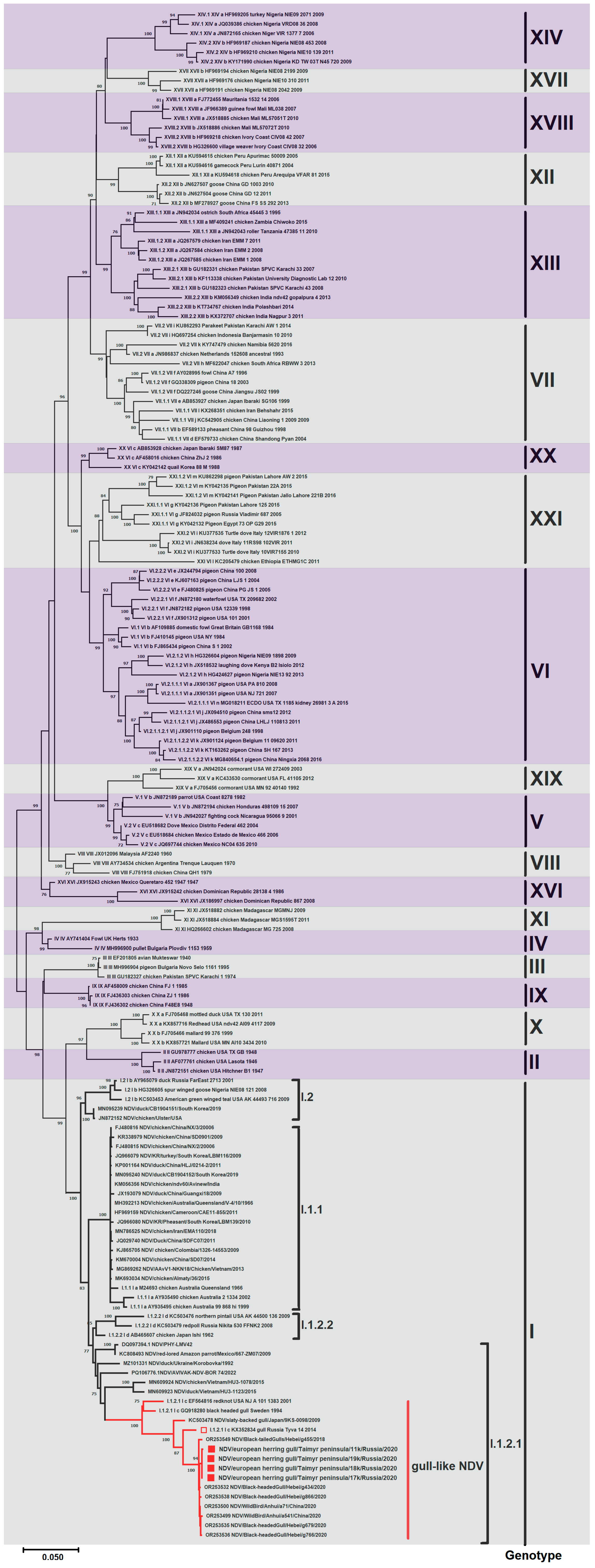
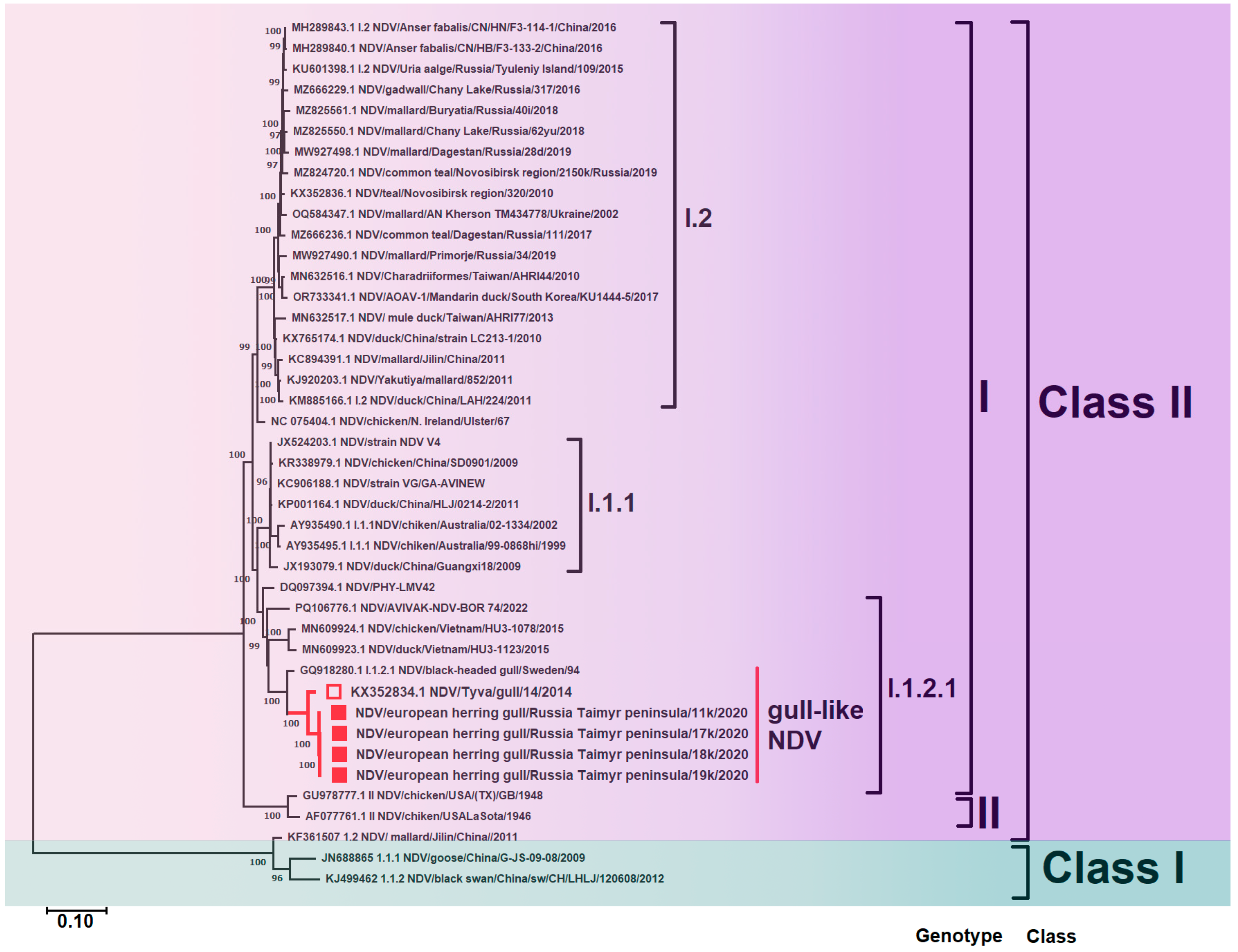
3.2. Phylogenetic Analysis of Newcastle Disease Virus
3.2.1. Gene F
3.2.2. Whole Genome
3.2.3. Amino Acid Substitutions
3.3. Phylogenetic Analysis of Avian Influenza Virus
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kitchen, A.; Shackelton, L.A.; Holmes, E.C. Family Level Phylogenies Reveal Modes of Macroevolution in RNA Viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 238–243. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, K.M.; Abolnik, C.; Afonso, C.L.; Albina, E.; Bahl, J.; Berg, M.; Briand, F.-X.; Brown, I.H.; Choi, K.-S.; Chvala, I.; et al. Updated Unified Phylogenetic Classification System and Revised Nomenclature for Newcastle Disease Virus. Infect. Genet. Evol. 2019, 74, 103917. [Google Scholar] [CrossRef]
- Alexander, D.J.; Aldous, E.W.; Fuller, C.M. The Long View: A Selective Review of 40 Years of Newcastle Disease Research. Avian Pathol. 2012, 41, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Sharshov, K.; Dubovitskiy, N.; Derko, A.; Loginova, A.; Kolotygin, I.; Zhirov, D.; Sobolev, I.; Kurskaya, O.; Alekseev, A.; Druzyaka, A.; et al. Does Avian Coronavirus Co-Circulate with Avian Paramyxovirus and Avian Influenza Virus in Wild Ducks in Siberia? Viruses 2023, 15, 1121. [Google Scholar] [CrossRef] [PubMed]
- Coffee, L.L.; Hanson, B.A.; Luttrell, M.P.; Swayne, D.E.; Senne, D.A.; Goekjian, V.H.; Niles, L.J.; Stallknecht, D.E. Avian Paramyxoviruses in Shorebirds and Gulls. J. Wildl. Dis. 2010, 46, 481–487. [Google Scholar] [CrossRef]
- Kim, L.M.; King, D.J.; Curry, P.E.; Suarez, D.L.; Swayne, D.E.; Stallknecht, D.E.; Slemons, R.D.; Pedersen, J.C.; Senne, D.A.; Winker, K.; et al. Phylogenetic Diversity among Low-Virulence Newcastle Disease Viruses from Waterfowl and Shorebirds and Comparison of Genotype Distributions to Those of Poultry-Origin Isolates. J. Virol. 2007, 81, 12641–12653. [Google Scholar] [CrossRef]
- Diel, D.G.; Miller, P.J.; Wolf, P.C.; Mickley, R.M.; Musante, A.R.; Emanueli, D.C.; Shively, K.J.; Pedersen, K.; Afonso, C.L. Characterization of Newcastle Disease Viruses Isolated from Cormorant and Gull Species in the United States in 2010. Avian Dis. 2012, 56, 128–133. [Google Scholar] [CrossRef]
- Kuiken, T.; Leighton, F.A.; Wobeser, G.; Danesik, K.L.; Riva, J.; Heckert, R.A. An Epidemic of Newcastle Disease in Double-Crested Cormorants from Saskatchewan. J. Wildl. Dis. 1998, 34, 457–471. [Google Scholar] [CrossRef]
- Relich, R.F.; Loeffelholz, M.J. Taxonomic Changes for Human Viruses, 2020 to 2022. J. Clin. Microbiol. 2022, 61, e0033722. [Google Scholar] [CrossRef]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Sreenivasan, C.C.; Li, F.; Wang, D. Emerging Threats of Highly Pathogenic Avian Influenza A (H5N1) in US Dairy Cattle: Understanding Cross-Species Transmission Dynamics in Mammalian Hosts. Viruses 2024, 16, 1703. [Google Scholar] [CrossRef] [PubMed]
- Echeverri-De la Hoz, D.; Martínez-Bravo, C.; Gastelbondo-Pastrana, B.; Rivero, R.; López, Y.; Bertel, V.; Alemán-Santos, M.; Garay, E.; Hoyos, R.; Arrieta, G.; et al. Genomics of Novel Influenza A Virus (H18N12) in Bats, Caribe Colombia. Sci. Rep. 2025, 15, 6507. [Google Scholar] [CrossRef] [PubMed]
- Gadzhiev, A.; Petherbridge, G.; Sharshov, K.; Sobolev, I.; Alekseev, A.; Gulyaeva, M.; Litvinov, K.; Boltunov, I.; Teymurov, A.; Zhigalin, A.; et al. Pinnipeds and Avian Influenza: A Global Timeline and Review of Research on the Impact of Highly Pathogenic Avian Influenza on Pinniped Populations with Particular Reference to the Endangered Caspian Seal (Pusa caspica). Front. Cell. Infect. Microbiol. 2024, 14, 1325977. [Google Scholar] [CrossRef]
- Dimitrov, K.M.; Ramey, A.M.; Qiu, X.; Bahl, J.; Afonso, C.L. Temporal, Geographic, and Host Distribution of Avian Paramyxovirus 1 (Newcastle Disease Virus). Infect. Genet. Evol. 2016, 39, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, A.M.; Chmura, A.A.; Gibbons, D.W.; Fleischer, R.C.; Marra, P.P.; Daszak, P. Predicting the Global Spread of H5N1 Avian Influenza. Proc. Natl. Acad. Sci. USA 2006, 103, 19368–19373. [Google Scholar] [CrossRef]
- Yoon, S.-W.; Webby, R.J.; Webster, R.G. Evolution and Ecology of Influenza A Viruses. In Influenza Pathogenesis and Control—Volume I; Compans, R.W., Oldstone, M.B.A., Eds.; Springer International Publishing: Cham, Switzerland, 2014; pp. 359–375. ISBN 978-3-319-11155-1. [Google Scholar]
- Boere, E.G.C.; Galbraith, C.A.; Stroud, D.A.; Thompson, D.B.A.; Underhill, L.G. Waterbirds Around the World; Stationery Office: Edinburgh, UK, 2006. [Google Scholar]
- Smeele, Z.E.; Ainley, D.G.; Varsani, A. Viruses Associated with Antarctic Wildlife: From Serology Based Detection to Identification of Genomes Using High Throughput Sequencing. Virus Res. 2018, 243, 91–105. [Google Scholar] [CrossRef]
- Banyard, A.C.; Bennison, A.; Byrne, A.M.P.; Reid, S.M.; Lynton-Jenkins, J.G.; Mollett, B.; De Silva, D.; Peers-Dent, J.; Finlayson, K.; Hall, R.; et al. Detection and Spread of High Pathogenicity Avian Influenza Virus H5N1 in the Antarctic Region. Nat. Commun. 2024, 15, 7433. [Google Scholar] [CrossRef]
- Ogrzewalska, M.; Vanstreels, R.T.; Pereira, E.; Campinas, E.; Junior, L.C.; Melo, J.O.; Macedo, L.; Appolinario, L.; Arantes, I.; Brandao, M.L.; et al. Genomic Analysis of High Pathogenicity Avian Influenza Viruses from Antarctica Reveals Multiple Introductions from South America. 12 June 2025. Preprint (Version 1). Available online: https://doi.org/10.21203/rs.3.rs-6727501/v1 (accessed on 28 June 2025).
- Kuiken, T.; Vanstreels, R.E.T.; Banyard, A.; Begeman, L.; Breed, A.C.; Dewar, M.; Fijn, R.; Serafini, P.P.; Uhart, M.; Wille, M. Emergence, Spread, and Impact of High-pathogenicity Avian Influenza H5 in Wild Birds and Mammals of South America and Antarctica. Conserv. Biol. 2025, 39, e70052. [Google Scholar] [CrossRef]
- Gass, J.D.; Kellogg, H.K.; Hill, N.J.; Puryear, W.B.; Nutter, F.B.; Runstadler, J.A. Epidemiology and Ecology of Influenza A Viruses among Wildlife in the Arctic. Viruses 2022, 14, 1531. [Google Scholar] [CrossRef]
- World Health Organization. World Health Organization Collecting, Preserving and Shipping Specimens for the Diagnosis of Avian Influenza A(H5N1) Virus Infection: Guide for Field Operations; World Health Organization: Geneva, Switzerland, 2006. [Google Scholar]
- Avian Influenza—WOAH—World Organisation for Animal Health. Available online: https://www.woah.org/en/disease/avian-influenza/ (accessed on 28 November 2024).
- Killian, M.L. Hemagglutination Assay for Influenza Virus. Methods Mol. Biol. 2014, 1161, 3–9. [Google Scholar] [CrossRef]
- van Boheemen, S.; Bestebroer, T.M.; Verhagen, J.H.; Osterhaus, A.D.M.E.; Pas, S.D.; Herfst, S.; Fouchier, R.A.M. A Family-Wide RT-PCR Assay for Detection of Paramyxoviruses and Application to a Large-Scale Surveillance Study. PLoS ONE 2012, 7, e34961. [Google Scholar] [CrossRef] [PubMed]
- Mine, J.; Tsunekuni, R.; Tanikawa, T.; Uchida, Y.; Dubovitskiy, N.; Derko, A.; Sobolev, I.; Shestopalov, A.; Sharshov, K.; Saito, T. Genetics of Japanese H5N8 High Pathogenicity Avian Influenza Viruses Isolated in Winter 2020–2021 and Their Genetic Relationship with Avian Influenza Viruses in Siberia. Transbound. Emerg. Dis. 2022, 69, e2195–e2213. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Donnelly, M.E.; Scholes, D.T.; George, K.S.; Hatta, M.; Kawaoka, Y.; Wentworth, D.E. Single-Reaction Genomic Amplification Accelerates Sequencing and Vaccine Production for Classical and Swine Origin Human Influenza A Viruses. J. Virol. 2009, 83, 10309–10313. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis Across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A Unified Bioinformatics Toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Postnikova, Y.; Treshchalina, A.; Gambaryan, A.; Belyakova, A.; Ishmukhametov, A.; Matrosovich, M.; Sadykova, G.; Prilipov, A.; Lomakina, N.; Boravleva, E. Evolutionary Dynamics of Avian Influenza Viruses Isolated from Wild Birds in Moscow. Int. J. Mol. Sci. 2023, 24, 3020. [Google Scholar] [CrossRef]
- Cui, H.; Shi, Y.; Ruan, T.; Li, X.; Teng, Q.; Chen, H.; Yang, J.; Liu, Q.; Li, Z. Phylogenetic Analysis and Pathogenicity of H3 Subtype Avian Influenza Viruses Isolated from Live Poultry Markets in China. Sci. Rep. 2016, 6, 27360. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, M.L.; Wise, H.M.; Nicol, M.Q.; Smith, N.; Dunfee, R.L.; Beard, P.M.; Jagger, B.W.; Ligertwood, Y.; Hardisty, G.R.; Xiao, H.; et al. Role of the B Allele of Influenza A Virus Segment 8 in Setting Mammalian Host Range and Pathogenicity. J. Virol. 2016, 90, 9263–9284. [Google Scholar] [CrossRef]
- Flusurver—Prepared for the Next Wave. Available online: https://flusurver.bii.a-star.edu.sg/ (accessed on 25 February 2025).
- Murashkina, T.; Sharshov, K.; Gadzhiev, A.; Petherbridge, G.; Derko, A.; Sobolev, I.; Dubovitskiy, N.; Loginova, A.; Kurskaya, O.; Kasianov, N.; et al. Avian Influenza Virus and Avian Paramyxoviruses in Wild Waterfowl of the Western Coast of the Caspian Sea (2017–2020). Viruses 2024, 16, 598. [Google Scholar] [CrossRef]
- Marco, M.A.D.; Sharshov, K.; Gulyaeva, M.; Delogu, M.; Ciccarese, L.; Castrucci, M.R.; Shestopalov, A. Ecology of Avian Influenza Viruses in Siberia; Nova Publishers: New York, NY, USA, 2016; Chapter 4. [Google Scholar]
- Druzyaka, A.V.; Druzyaka, O.R.; Sharshov, K.A.; Kasianov, N.; Dubovitskiy, N.; Derko, A.A.; Frolov, I.G.; Torniainen, J.; Wang, W.; Minina, M.A.; et al. Stable Isotope Analysis Reveals Common Teal (Anas crecca) Molting Sites in Western Siberia: Implications for Avian Influenza Virus Spread. Microorganisms 2024, 12, 357. [Google Scholar] [CrossRef]
- Sun, H.; Li, H.; Tong, Q.; Han, Q.; Liu, J.; Yu, H.; Song, H.; Qi, J.; Li, J.; Yang, J.; et al. Airborne Transmission of Human-Isolated Avian H3N8 Influenza Virus Between Ferrets. Cell 2023, 186, 4074–4084.e11. [Google Scholar] [CrossRef]
- Austin, F.J.; Webster, R.G. Evidence of Ortho- and Paramyxoviruses in Fauna from Antarctica. J. Wildl. Dis. 1993, 29, 568–571. [Google Scholar] [CrossRef]
- Morgan, I.R.; Westbury, H.A. Studies of Viruses in Penguins in the Vestfold Hills. Hydrobiologia 1988, 165, 263–269. [Google Scholar] [CrossRef]
- Thomazelli, L.M.; Araujo, J.; Oliveira, D.B.; Sanfilippo, L.; Ferreira, C.S.; Brentano, L.; Pelizari, V.H.; Nakayama, C.; Duarte, R.; Hurtado, R.; et al. Newcastle Disease Virus in Penguins from King George Island on the Antarctic Region. Vet. Microbiol. 2010, 146, 155–160. [Google Scholar] [CrossRef]
- Sivay, M.V.; Silko, N.Y.; Sharshov, K.A.; Prokudin, A.V.; Li, L.; Yang, M.; Cao, S.; Shestopalov, A.M. The Role of Wild Goose (Anser) Populations of Russia and the Tibet Plateau in the Spread of the Avian Influenza Virus. Chin. Birds 2011, 2, 140–146. [Google Scholar] [CrossRef]
- Munir, M.; Linde, A.-M.; Zohari, S.; Ståhl, K.; Baule, C.; Holm, K.; Engström, B.; Berg, M. Complete Genome Analysis of an Avian Paramyxovirus Type 1 Strain Isolated in 1994 from an Asymptomatic Black-Headed Gull (Larus ridibundus) in Southern Sweden. Avian Dis. 2010, 54, 923–930. [Google Scholar] [CrossRef]
- Ramey, A.M.; Reeves, A.B.; Ogawa, H.; Ip, H.S.; Imai, K.; Bui, V.N.; Yamaguchi, E.; Silko, N.Y.; Afonso, C.L. Genetic Diversity and Mutation of Avian Paramyxovirus Serotype 1 (Newcastle Disease Virus) in Wild Birds and Evidence for Intercontinental Spread. Arch. Virol. 2013, 158, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Larus [Vegae or Mongolicus] (Vega or Mongolian Gull)—Avibase. Available online: https://avibase.bsc-eoc.org/species.jsp?lang=EN&avibaseid=F01A756B334DF946 (accessed on 15 February 2025).
- Birds of Russia. Herring Gull/Larus Argentatus (Pontoppidan, 1763). Available online: https://www.egir.ru/bird/87.html (accessed on 15 February 2025).
- World Organisation for Animal Health (WOAH), Newcastle Disease (Infection with Newcastle Disease Virus). WOAH Terrestrial Manual 2021; World Organisation for Animal Health: Paris, France, 2021; Chapter 3.3.14. [Google Scholar]
- Reid, S.M.; Byrne, A.M.P.; Lean, F.Z.X.; Ross, C.S.; Pascu, A.; Hepple, R.; Dominguez, M.; Frost, S.; Coward, V.J.; Núñez, A.; et al. A Multi-Species, Multi-Pathogen Avian Viral Disease Outbreak Event: Investigating Potential for Virus Transmission at the Wild Bird—Poultry Interface. Emerg. Microbes Infect. 2024, 13, 2348521. [Google Scholar] [CrossRef] [PubMed]
- Wille, M.; Avril, A.; Tolf, C.; Schager, A.; Larsson, S.; Borg, O.; Olsen, B.; Waldenström, J. Temporal Dynamics, Diversity, and Interplay in Three Components of the Virodiversity of a Mallard Population: Influenza A Virus, Avian Paramyxovirus and Avian Coronavirus. Infect. Genet. Evol. 2015, 29, 129–137. [Google Scholar] [CrossRef]
- Kattenbelt, J.A.; Stevens, M.P.; Selleck, P.W.; Gould, A.R. Analysis of Newcastle Disease Virus Quasispecies and Factors Affecting the Emergence of Virulent Virus. Arch. Virol. 2010, 155, 1607–1615. [Google Scholar] [CrossRef]
- Yamaguchi, N.; Hupp, J.W.; Higuchi, H.; Flint, P.L.; Pearce, J.M. Satellite-tracking of Northern Pintail Anas Acuta during Outbreaks of the H5N1 Virus in Japan: Implications for Virus Spread. Ibis 2010, 152, 262–271. [Google Scholar] [CrossRef]
- Jahangir, A.; Watanabe, Y.; Chinen, O.; Yamazaki, S.; Sakai, K.; Okamura, M.; Nakamura, M.; Takehara, K. Surveillance of Avian Influenza Viruses in Northern Pintails (Anas acuta) in Tohoku District, Japan. Avian Dis. 2008, 52, 49–53. [Google Scholar] [CrossRef]
- Jahangir, A.; Ruenphet, S.; Ueda, S.; Ueno, Y.; Shoham, D.; Shindo, J.; Okamura, M.; Nakamura, M.; Takehara, K. Avian Influenza and Newcastle Disease Viruses from Northern Pintail in Japan: Isolation, Characterization and Inter-Annual Comparisons during 2006–2008. Virus Res. 2009, 143, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Uhart, M.M.; Vanstreels, R.E.T.; Nelson, M.I.; Olivera, V.; Campagna, J.; Zavattieri, V.; Lemey, P.; Campagna, C.; Falabella, V.; Rimondi, A. Epidemiological Data of an Influenza A/H5N1 Outbreak in Elephant Seals in Argentina Indicates Mammal-to-Mammal Transmission. Nat. Commun. 2024, 15, 9516. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Family | Species | Total Number | Number of NDV | Number of AIV |
|---|---|---|---|---|---|
| Anseriformes | Anatidae | Greater white-fronted goose (Anser albifrons) | 9 | 0 | 0 |
| 12 species (n = 147) | Anatidae | Common merganser (Mergus merganser) | 4 | 0 | 0 |
| Anatidae | Long-tailed duck (Clangula hyemalis) | 15 | 0 | 0 | |
| Anatidae | Common goldeneye (Bucephala clangula) | 7 | 0 | 0 | |
| Anatidae | Taiga bean-goose (Anser fabalis) | 1 | 0 | 0 | |
| Anatidae | Eurasian wigeon (Mareca penelope) | 28 | 0 | 0 | |
| Anatidae | Velvet scoter (Melanitta fusca) | 28 | 0 | 0 | |
| Anatidae | Gadwall (Mareca strepera) | 1 | 0 | 0 | |
| Anatidae | Greater scaup (Aythya marila) | 18 | 0 | 0 | |
| Anatidae | Tufted duck (Aythya fuligula) | 2 | 0 | 0 | |
| Anatidae | Green-winged teal (Anas crecca) | 4 | 0 | 0 | |
| Anatidae | Northern pintail (Anas acuta) | 30 | 0 | 1 | |
| Charadriiformes | Stercorariidae | Long-tailed jaeger (Stercorarius longicaudus) | 2 | 0 | 0 |
| 4 species (n = 167) | Charadriidae | European golden-plover (Pluvialis apricaria) | 4 | 0 | 0 |
| Scolopacidae | Ruff (Calidris pugnax) | 3 | 0 | 0 | |
| Laridae | Herring gull (Larus argentatus) | 158 | 4 | 0 | |
| Gaviiformes 1 species (n = 7) | Gaviidae | Arctic loon (Gavia arctica) | 7 | 0 | 0 |
| Passeriformes 1 species (n = 2) | Corvidae | Hooded crow (Corvus cornix) | 2 | 0 | 0 |
| Total | 7 | 18 | 323 | 4 | 1 |
| Strain | Host | Related Strain | Country | Host | Whole-Genome Identity, % | Gene F Identity, % |
|---|---|---|---|---|---|---|
| NDV/11k | Larus argentatus | APMV-1/WildBird/Anhui/a71/2020 | China | Wild bird | - 1 | 99.76 |
| APMV-1/Black-headedGull/Hebei/g679/2020 | China | Chroicocephalus ridibundus | - | 99.70 | ||
| NDV/Tyva/gull/14/2014 | Russia | Hydrocoloeus minutus | 97.30 | 97.29 | ||
| APMV-1/slaty-backed gull/Japan/9KS-0098/2009 | Japan | Larus schistisagus | - | 96.63 | ||
| APMV-1/red knot/US(NJ)/A101-1383/2001 | USA | Calidris canutus | - | 94.04 | ||
| NDV/BHG/Sweden/94 | Sweden | Chroicocephalus ridibundus | 94.44 | 93.98 | ||
| PHY-LMV42 | Hungary | Vaccine strain | 91.41 | 91.16 | ||
| AVIVAK-NDV-BOR 74 | Russia | Vaccine strain | 90.72 | 91.82 | ||
| NDV/chicken/Vietnam/HU3-1078/2015 | Vietnam | Chicken | 90.31 | 90.55 | ||
| NDV/17k | Larus argentatus | APMV-1/WildBird/Anhui/a71/2020 | China | Wild bird | - | 99.82 |
| APMV-1/Black-headedGull/Hebei/g679/2020 | China | Chroicocephalus ridibundus | - | 99.76 | ||
| NDV/Tyva/gull/14/2014 | Russia | Hydrocoloeus minutus | 97.24 | 97.35 | ||
| APMV-1/slaty-backed gull/Japan/9KS-0098/2009 | Japan | Larus schistisagus | - | 96.69 | ||
| APMV-1/red knot/US(NJ)/A101-1383/2001 | USA | Calidris canutus | - | 94.10 | ||
| NDV/BHG/Sweden/94 | Sweden | Chroicocephalus ridibundus | 94.40 | 94.04 | ||
| PHY-LMV42 | Hungary | Vaccine strain | 91.37 | 91.16 | ||
| AVIVAK-NDV-BOR 74 | Russia | Vaccine strain | 90.68 | 91.88 | ||
| NDV/chicken/Vietnam/HU3-1078/2015 | Vietnam | Chicken | 90.30 | 90.61 | ||
| NDV/18k | Larus argentatus | APMV-1/WildBird/Anhui/a71/2020 | China | Wild bird | - | 99.82 |
| APMV-1/Black-headedGull/Hebei/g679/2020 | China | Chroicocephalus ridibundus | - | 99.76 | ||
| NDV/Tyva/gull/14/2014 | Russia | Hydrocoloeus minutus | 97.23 | 97.35 | ||
| APMV-1/slaty-backed gull/Japan/9KS-0098/2009 | Japan | Larus schistisagus | - | 96.69 | ||
| APMV-1/red knot/US(NJ)/A101-1383/2001 | USA | Calidris canutus | - | 94.10 | ||
| NDV/BHG/Sweden/94 | Sweden | Chroicocephalus ridibundus | 94.39 | 94.04 | ||
| PHY-LMV42 | Hungary | Vaccine strain | 91.36 | 91.22 | ||
| AVIVAK-NDV-BOR 74 | Russia | Vaccine strain | 90.68 | 91.88 | ||
| NDV/chicken/Vietnam/HU3-1078/2015 | Vietnam | Chicken | 90.29 | 90.55 | ||
| NDV/19k | Larus argentatus | APMV-1/WildBird/Anhui/a71/2020 | China | Wild bird | - | 99.82 |
| APMV-1/Black-headedGull/Hebei/g679/2020 | China | Chroicocephalus ridibundus | - | 99.76 | ||
| NDV/Tyva/gull/14/2014 | Russia | Hydrocoloeus minutus | 97.22 | 97.35 | ||
| APMV-1/slaty-backed gull/Japan/9KS-0098/2009 | Japan | Larus schistisagus | - | 96.69 | ||
| APMV-1/red knot/US(NJ)/A101-1383/2001 | USA | Calidris canutus | - | 94.10 | ||
| NDV/BHG/Sweden/94 | Sweden | Chroicocephalus ridibundus | 94.39 | 94.04 | ||
| PHY-LMV42 | Hungary | Vaccine strain | 91.35 | 91.22 | ||
| AVIVAK-NDV-BOR 74 | Russia | Vaccine strain | 90.67 | 91.88 | ||
| NDV/chicken/Vietnam/HU3-1078/2015 | Vietnam | Chicken | 90.29 | 90.61 |
| Strain | Cleavage Site Amino acid Sequence | Year of Isolation | Sub-Genotype | GenBank Accession No. |
|---|---|---|---|---|
| BHG/Sweden/94 | 112GKQGR↓L117 | 1994 | I.1.2.1 | GQ918280 |
| Slaty-backed gull/Japan/9KS-0098/2009 | 2009 | I.1.2.1 | KC503478 | |
| Tyva/gull/14/2014 | 2014 | I.1.2.1 | KX352834 | |
| Black-headedGull/Hebei/g679/2020 | 2020 | I.1.2.1 | OR253535 | |
| WildBird/Anhui/a71/2020 | 2020 | I.1.2.1 | OR253500 | |
| Red knot/USA/NJ/A101/1383/2001 | 2001 | I.1.2.1 | EF564816 | |
| NDV/11k | 2020 | I.1.2.1 | PV032627 | |
| NDV/17k | 2020 | I.1.2.1 | PV032628 | |
| NDV/18k | 2020 | I.1.2.1 | PV032629 | |
| NDV/19k | 2020 | I.1.2.1 | PV032630 | |
| NDV/chicken/Vietnam/HU3-1078/2015 | 2015 | I.1.2.1 | MN609924 | |
| AVIVAK-NDV-BOR 74 | 2022 | I.1.2.1 | PQ106776 | |
| PHY-LMV42 | I.1.2.1 | DQ097394 | ||
| Northern pintail/USA/AK/44500/136/2009 | 112EKQGR↓L117 | 2009 | I.1.2.2 | KC503476 |
| Chicken/Japan/Ishi/1962 | 112GKQGR↓L117 | 1962 | I.1.2.2 | AB465607 |
| Chicken/Australia/Queensland/V-4/10/1966 | 112GKQGR↓L117 | 1966 | I.1.1 | MH392213 |
| Chicken/Australia/2/1334/2002 | 112RRQRR↓F117 | 2002 | I.1.1 | AY935490 |
| Chicken/Australia99/86 hi/1999 | 112RRQGR↓L117 | 1999 | I.1.1 | AY935495 |
| Chicken/Ulster/USA | 112GKQGR↓L117 | 1967 | I.2 | JN872152 |
| Duck/Russia/FarEast/2713/2001 | 2001 | I.2 | AY965079 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Derko, A.; Dubovitskiy, N.; Prokudin, A.; Mine, J.; Tsunekuni, R.; Uchida, Y.; Saito, T.; Kasianov, N.; Loginova, A.; Sobolev, I.; et al. Detection of a Novel Gull-like Clade of Newcastle Disease Virus and H3N8 Avian Influenza Virus in the Arctic Region of Russia (Taimyr Peninsula). Viruses 2025, 17, 955. https://doi.org/10.3390/v17070955
Derko A, Dubovitskiy N, Prokudin A, Mine J, Tsunekuni R, Uchida Y, Saito T, Kasianov N, Loginova A, Sobolev I, et al. Detection of a Novel Gull-like Clade of Newcastle Disease Virus and H3N8 Avian Influenza Virus in the Arctic Region of Russia (Taimyr Peninsula). Viruses. 2025; 17(7):955. https://doi.org/10.3390/v17070955
Chicago/Turabian StyleDerko, Anastasiya, Nikita Dubovitskiy, Alexander Prokudin, Junki Mine, Ryota Tsunekuni, Yuko Uchida, Takehiko Saito, Nikita Kasianov, Arina Loginova, Ivan Sobolev, and et al. 2025. "Detection of a Novel Gull-like Clade of Newcastle Disease Virus and H3N8 Avian Influenza Virus in the Arctic Region of Russia (Taimyr Peninsula)" Viruses 17, no. 7: 955. https://doi.org/10.3390/v17070955
APA StyleDerko, A., Dubovitskiy, N., Prokudin, A., Mine, J., Tsunekuni, R., Uchida, Y., Saito, T., Kasianov, N., Loginova, A., Sobolev, I., Kumar, S., Shestopalov, A., & Sharshov, K. (2025). Detection of a Novel Gull-like Clade of Newcastle Disease Virus and H3N8 Avian Influenza Virus in the Arctic Region of Russia (Taimyr Peninsula). Viruses, 17(7), 955. https://doi.org/10.3390/v17070955