


Viruses 2026, 18(6), 658; https://doi.org/10.3390/v18060658 (registering DOI) - 9 Jun 2026
Abstract
DNA viruses rely extensively on host cellular machinery, including replication factors and transcriptional systems, to persist after infection. These mechanisms make studying and targeting DNA viral proteins challenging, as they also play key roles in mammalian processes. Traditional strategies include CRISPR-mediated gene disruption
[...] Read more.
DNA viruses rely extensively on host cellular machinery, including replication factors and transcriptional systems, to persist after infection. These mechanisms make studying and targeting DNA viral proteins challenging, as they also play key roles in mammalian processes. Traditional strategies include CRISPR-mediated gene disruption and small interfering RNA (siRNA) to target host proteins. However, Proteolysis Targeting Chimeras (PROTACs) offer a novel strategy by enabling the selective and rapid degradation of specific viral or host proteins involved in the DNA viral lifecycle. PROTACs are heterobifunctional molecules composed of three key components: a ligand that binds the target protein, a chemical linker, and a ligand that recruits an E3 ubiquitin ligase. By simultaneously binding both the target protein and the E3 ligase, PROTACs form a ternary complex. This proximity enables the E3 ligase to ubiquitinate the target protein, marking it for recognition and subsequent degradation by the intracellular proteasome. This approach represents a promising avenue for targeting previously undruggable proteins and improving therapeutic outcomes in virus-associated malignancies. In this perspective, we describe studies that use PROTACs as tools to modulate host proteins to investigate DNA viral processes with temporal control of host protein expression, as well as the use of PROTACs as antivirals to directly target DNA viral proteins. We also provide a detailed chart summarizing known host-targeting PROTACs and their potential applications across different stages of DNA viral lifecycles, highlighting opportunities for future DNA virus research.
Full article
(This article belongs to the Section Human Virology and Viral Diseases)
►
Show Figures
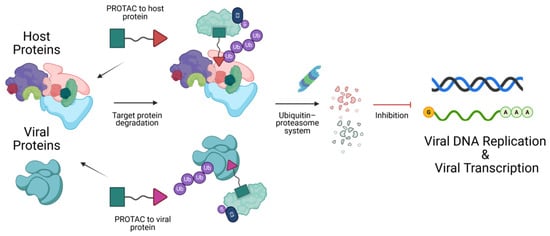
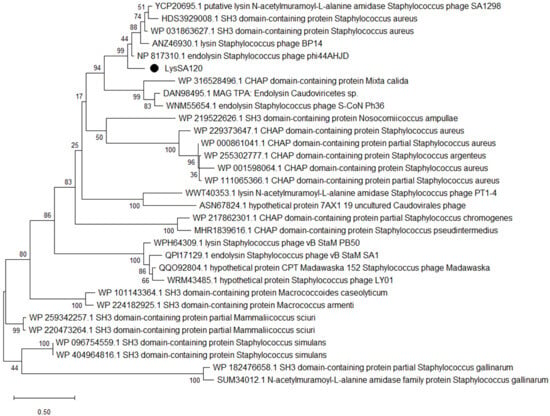
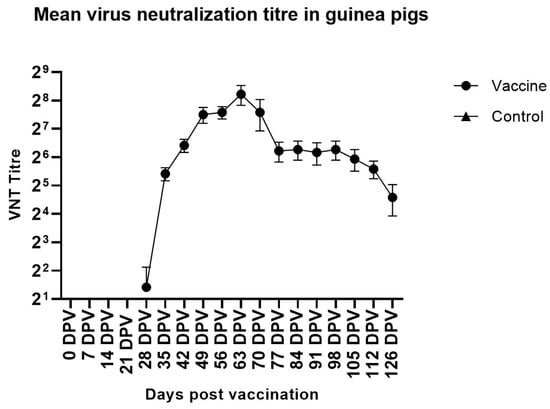
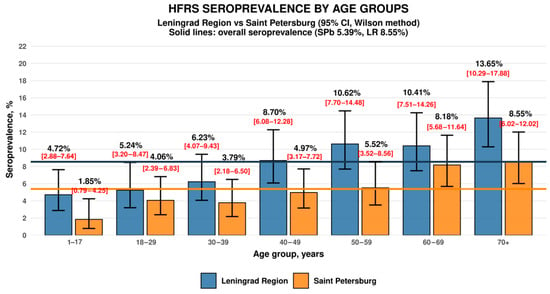
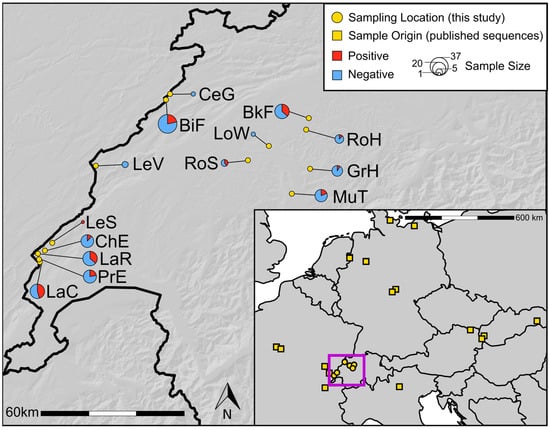
Figure 1










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}