


Viruses 2026, 18(7), 694; https://doi.org/10.3390/v18070694 (registering DOI) - 24 Jun 2026
Abstract
Oncolytic viruses represent a promising class of anticancer therapeutics, and rapid, accurate quantification of viral titers is critical for ensuring both efficacy and safety during clinical development. Conventional viral titering methods, such as 50% cell culture infectious dose (CCID50), are time-consuming
[...] Read more.
Oncolytic viruses represent a promising class of anticancer therapeutics, and rapid, accurate quantification of viral titers is critical for ensuring both efficacy and safety during clinical development. Conventional viral titering methods, such as 50% cell culture infectious dose (CCID50), are time-consuming and limited in sensitivity, thereby restricting their application in real-time clinical monitoring. This study aimed to develop and validate a rapid titer assay for oHSV2-PD-L1/CD3-BsAb, an oncolytic herpes simplex virus expressing a PD-L1/CD3 bispecific antibody, to support preclinical and clinical monitoring. A dual-reporter cell system was established using Vero-PD-L1-GFP (Vero cells expressing PD-L1 and GFP) cells as target cells and Jurkat-NFAT-Fluc (Jurkat cells expressing NFAT and Fluc) cells as effector cells. Viral infection activates the NFAT signaling pathway, driving Fluc expression, thereby enabling rapid quantification of infectious virus. The assay was evaluated for specificity, limit of detection (LOD), and lower limit of quantification (LLOQ), and compared with the conventional CCID50 method. Its applicability was further assessed using clinical simulation samples, including PBMCs and swabs. The rapid titer assay accurately quantified virus at 103 CCID50/mL after 8 h of incubation, consistent with CCID50 results, while extending the incubation to 18 h improved the LLOQ to 102.5 CCID50/mL, demonstrating enhanced sensitivity. The assay exhibited high reproducibility and stability in both PBMC and swab samples, enabling reliable quantification of low-titer virus in complex biological matrices. Compared with CCID50, the method substantially reduced assay time (from 3–5 days to 8–18 h) while improving sensitivity and specificity. The developed rapid titer assay for oHSV2-PD-L1/CD3-BsAb provides a sensitive and specific platform for viral quantification. It offers a valuable tool for oncolytic virus development, production quality control, and clinical monitoring, facilitating efficient safety evaluation and risk management in ongoing and future clinical applications.
Full article
(This article belongs to the Section Human Virology and Viral Diseases)
►
Show Figures
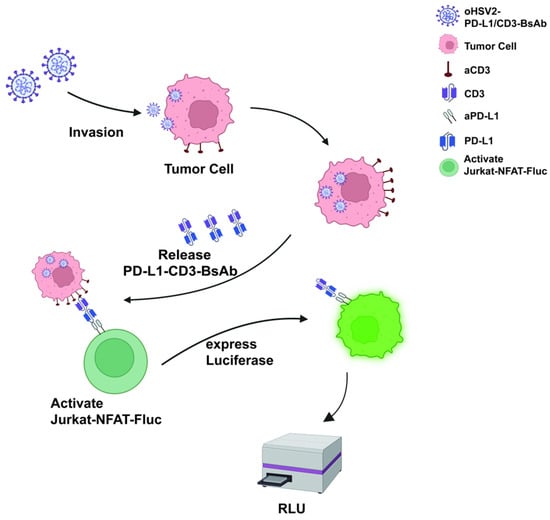
Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}