




Non-Coding RNA 2026, 12(4), 25; https://doi.org/10.3390/ncrna12040025 - 22 Jul 2026
Abstract
Rett syndrome (RTT) is a severe X-linked neurodevelopmental disorder that is caused in most cases by pathogenic variants in MECP2, the gene encoding methyl-CpG-binding protein 2 (MeCP2). Despite substantial progress in the development of gene therapy, restoring MECP2 expression remains challenging because
[...] Read more.
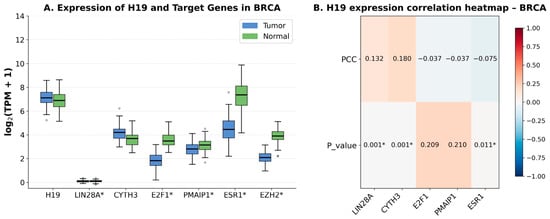
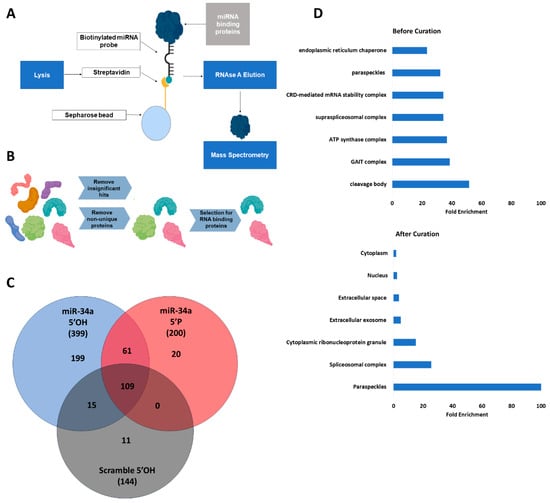
Rett syndrome (RTT) is a severe X-linked neurodevelopmental disorder that is caused in most cases by pathogenic variants in MECP2, the gene encoding methyl-CpG-binding protein 2 (MeCP2). Despite substantial progress in the development of gene therapy, restoring MECP2 expression remains challenging because MeCP2 is highly dosage-sensitive. Both deficiency and excessive expression of this protein are associated with severe neurological abnormalities. This makes simple viral vector-mediated replacement of MECP2 potentially unsafe and underscores the need for multilayered systems that control transgene expression. This review discusses current and emerging strategies for regulating MeCP2 expression in RTT, with an emphasis on non-coding RNA-based and epigenetic mechanisms. Particular attention is given to the limitations of conventional AAV-mediated gene therapy, the use of cell-specific and endogenous promoters, miRNA-regulated elements, autoregulatory systems, and post-transcriptional control of MECP2 expression. Strategies for reactivating the inactive X chromosome are also discussed, including XIST-dependent regulation and epigenome editing. In addition, the review considers CRISPR-mediated regulation, selective epigenetic activation, and combined therapeutic platforms that integrate viral delivery, RNA-dependent post-transcriptional control, and endogenous gene regulation. Overall, clinically applicable gene therapy for RTT will likely need to move beyond simple MECP2 replacement and instead rely on precise cell- and dose-dependent regulation of its expression. Non-coding RNA and epigenetic mechanisms represent important layers of such control and may contribute to the development of safer gene therapy strategies for RTT.
Full article
(This article belongs to the Section Clinical Applications of Non-Coding RNA)
►
Show Figures

Figure 1




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}