







Genes 2025, 16(5), 561; https://doi.org/10.3390/genes16050561 - 8 May 2025
Abstract
Background: Asilomorpha, an infraorder of predatory Diptera (Brachycera), is of significant evolutionary interest due to their remarkable ecological diversity, broad size range, and specialized feeding behaviors. However, phylogenetic studies of this group have been limited by sampling challenges. Methods: In this study, we
[...] Read more.
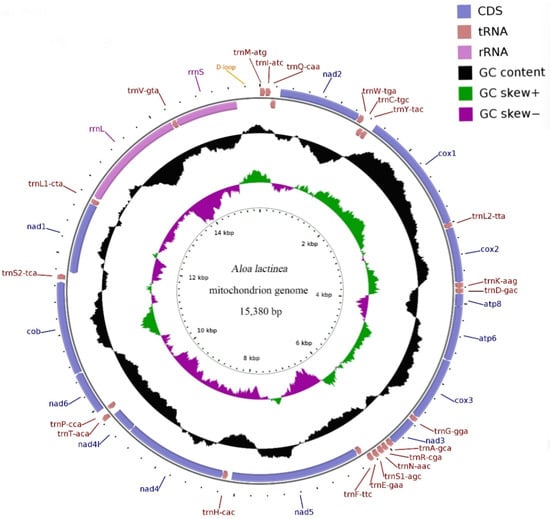
Background: Asilomorpha, an infraorder of predatory Diptera (Brachycera), is of significant evolutionary interest due to their remarkable ecological diversity, broad size range, and specialized feeding behaviors. However, phylogenetic studies of this group have been limited by sampling challenges. Methods: In this study, we sequenced the complete mitochondrial genomes of two Chinese endemic species, Clephydroneura jiangxiensis (C. jiangxiensis) and Maira xizangensis (M. xizangensis), using whole-genome random sequencing. By integrating these novel data with published sequences from NCBI, we reconstructed the phylogeny of three superfamilies (Asiloidea, Empidoidea, and Nemestrinoidea). Results: Both mitochondrial genomes exhibit the typical 37 genes (13 protein-coding genes, 22 tRNAs, and 2 rRNAs) and display pronounced AT bias. Congruent results from maximum likelihood analysis and Bayesian inference strongly supported the ideas that both new species are placed in Asilidae and that the Asilidae family is monophyletic. However, relationships among the three superfamilies remain unclear. Our results suggest that (1) although Asiloidea and Nemestrinidea are closely related, the potential positioning of Nemestrinoidea as an independent superfamily is worth investigating; and (2) Empidoidea may form a sister group to Asiloidea + Nemestrinidae, though this hypothesis requires further corroboration given the basal position of Hemipenthes hebeiensis (Bombyliidae). Conclusions: These findings highlight the need for expanded taxon sampling, particularly of underrepresented families, to resolve deep-level relationships within Asilomorpha. Clarifying the phylogenetic relationships within Asilomorpha will facilitate future investigations into their evolutionary origins and the evolution of characteristic traits.
Full article
(This article belongs to the Special Issue Molecular Evolution, Mitochondrial Genomics and Mitochondrial Genome Expression in Animals: 2024–2025)
►
Show Figures

Figure 1









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}