



Genes 2026, 17(4), 386; https://doi.org/10.3390/genes17040386 - 28 Mar 2026
Viewed by 524
Abstract
►
Show Figures
Background: The WRKY transcription factor family represents one of the most crucial transcription factor families in plants, regulating diverse physiological processes. The heartwood of Dalbergia odorifera is a prized material for both high-quality rosewood and traditional medicinal applications, exhibiting exceptional economic value. However,
[...] Read more.
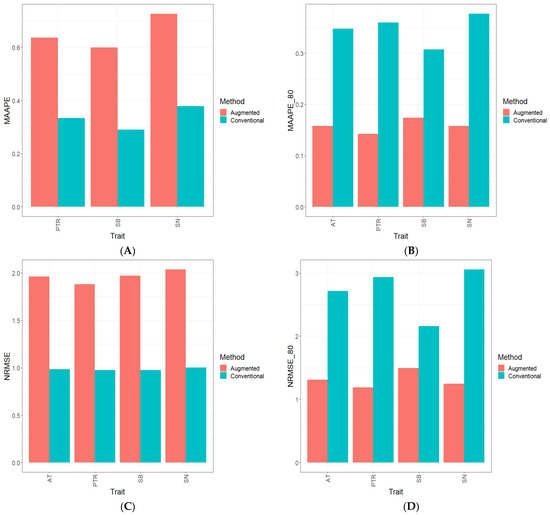
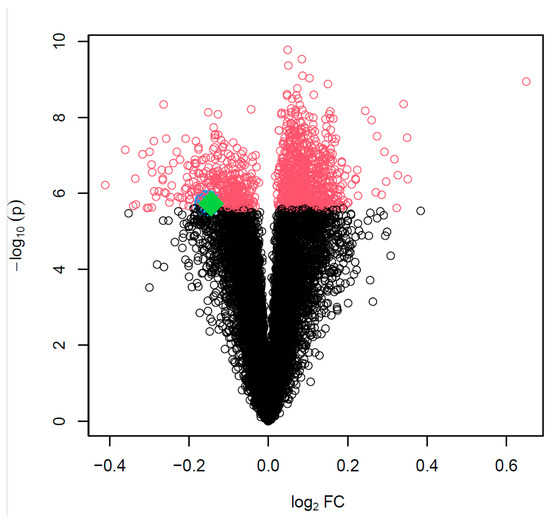
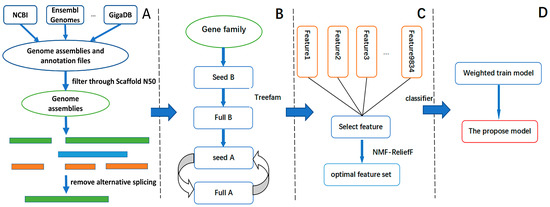
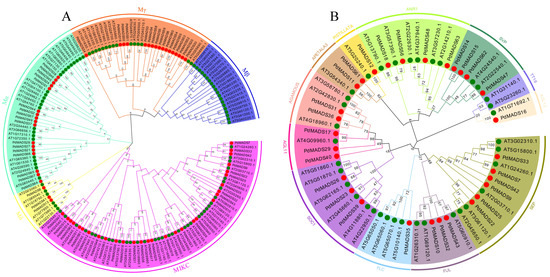
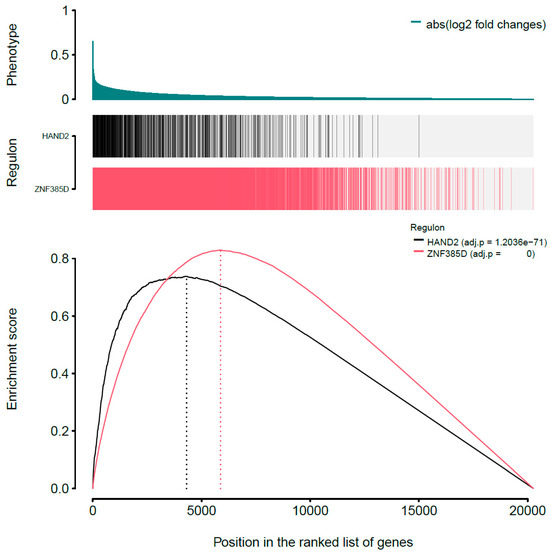
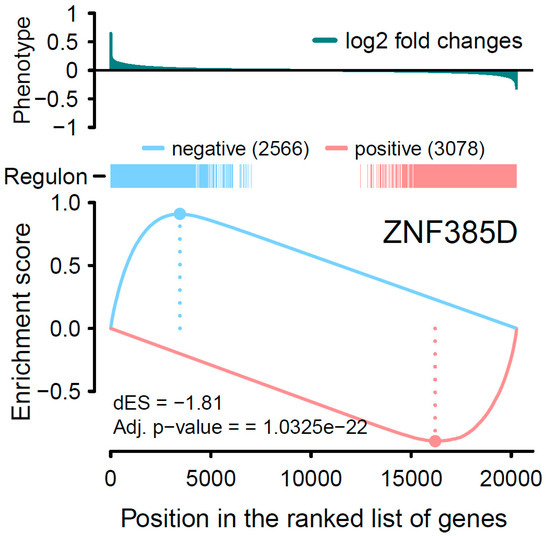
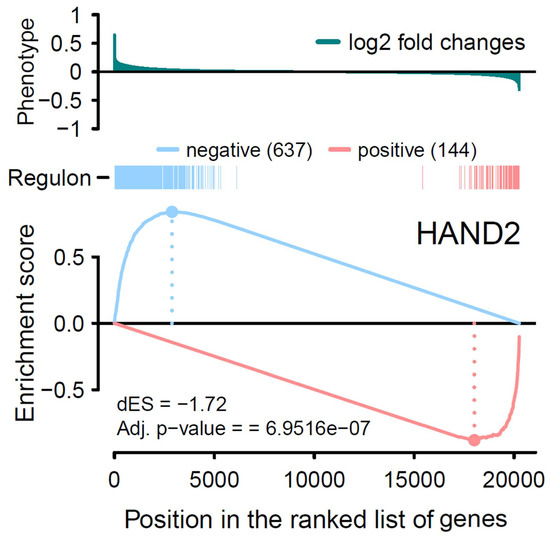
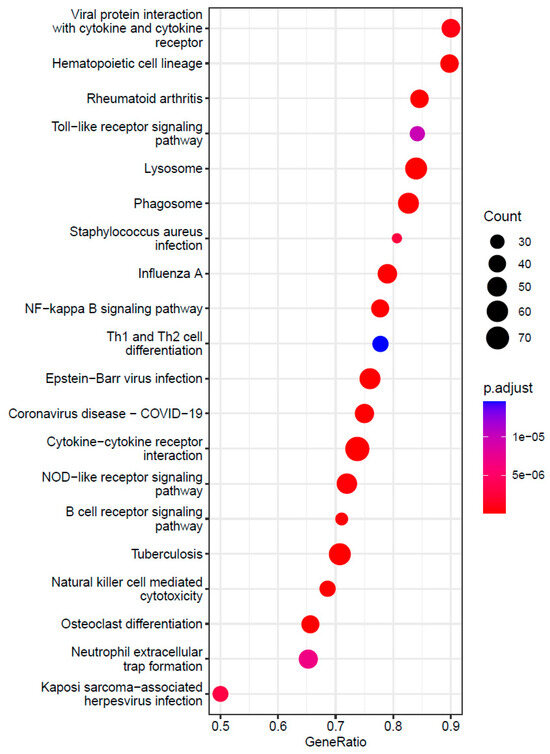
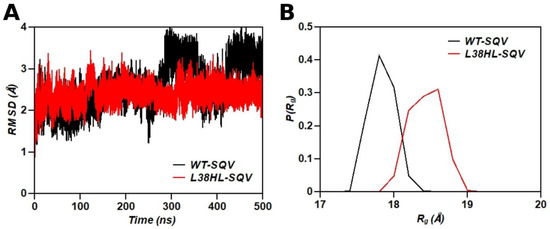
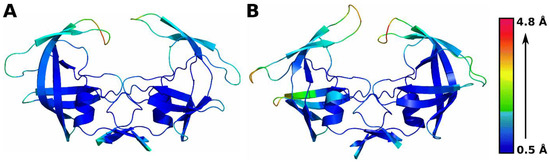
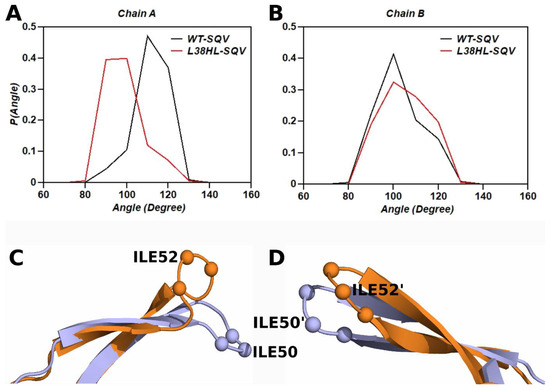
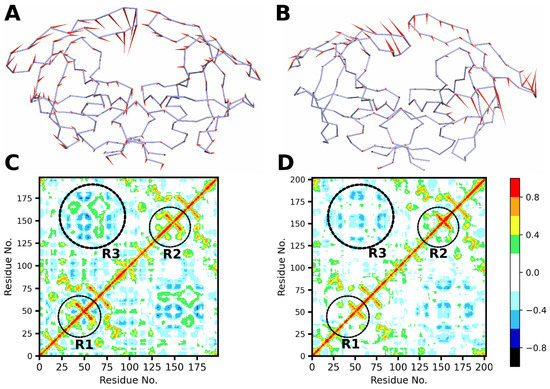
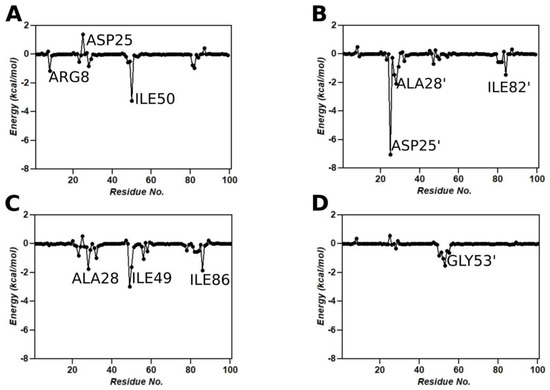
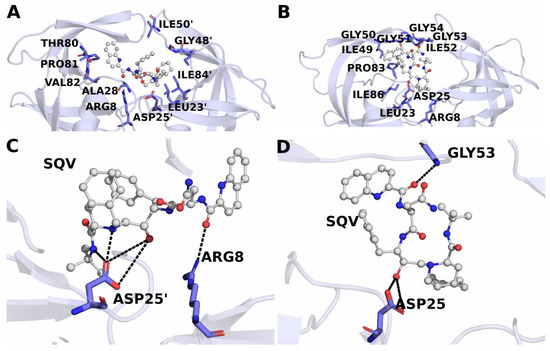
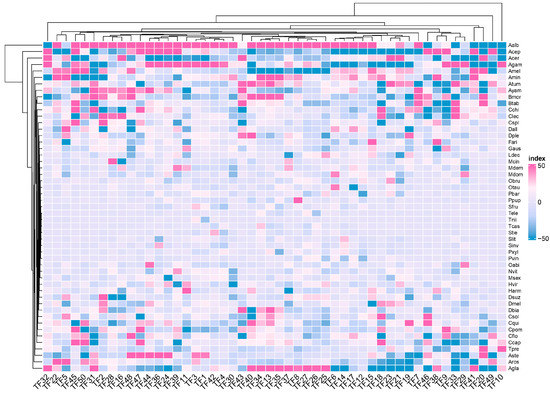
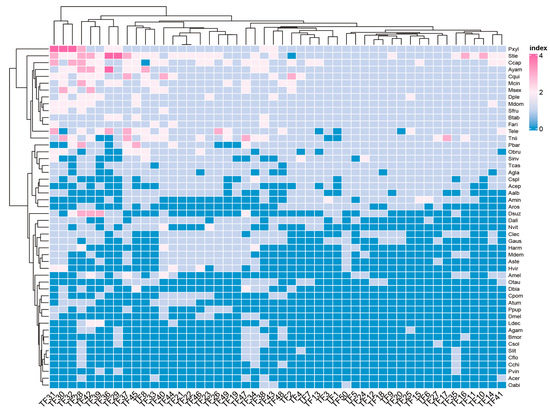
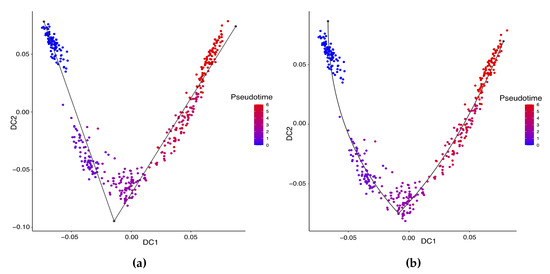
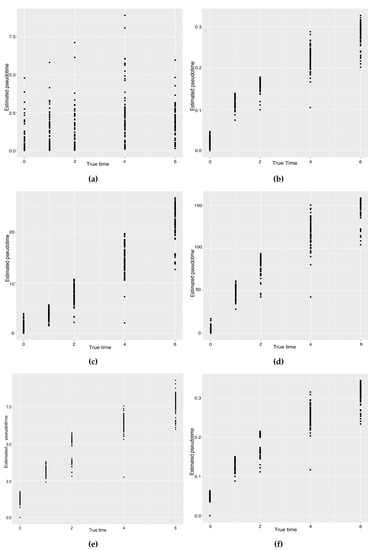
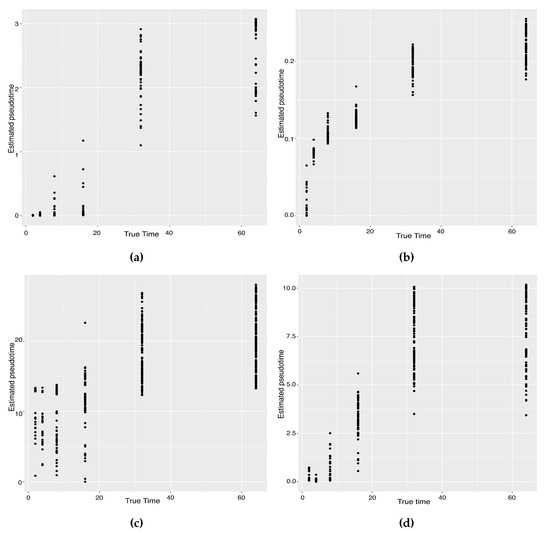
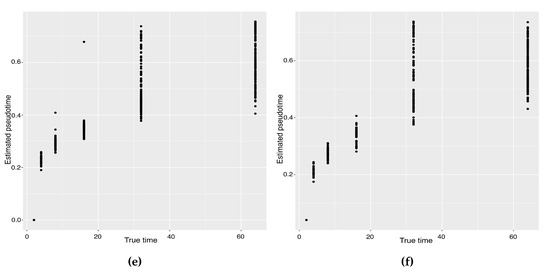
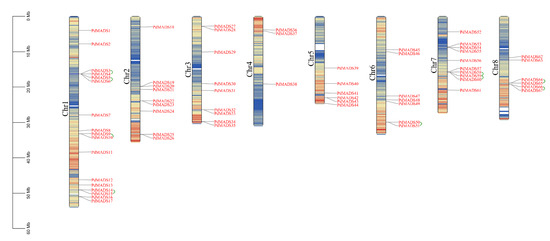
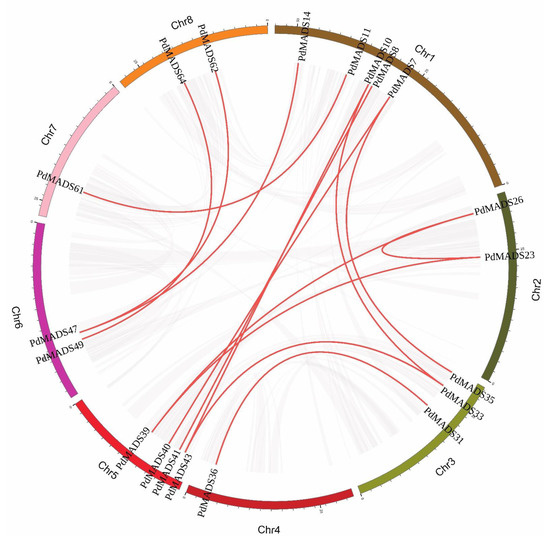
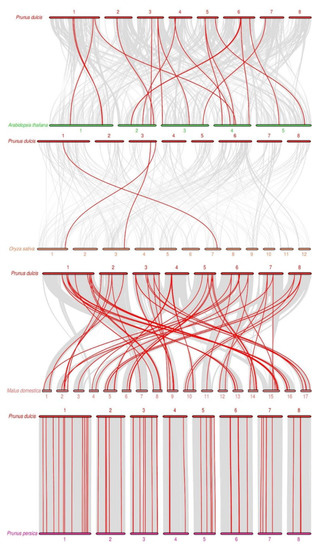
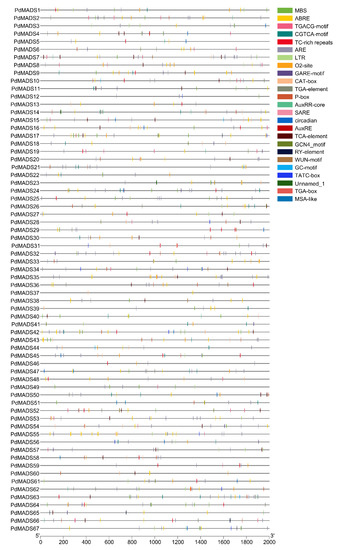
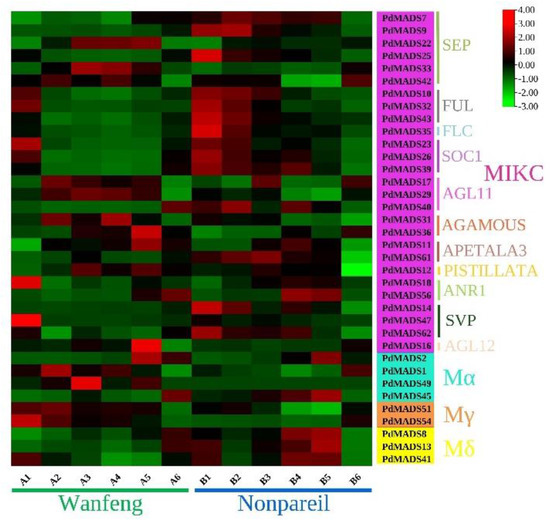
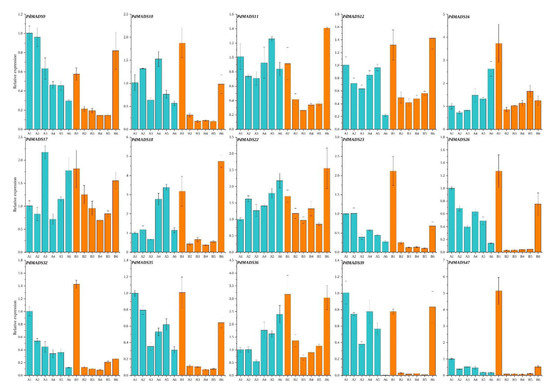
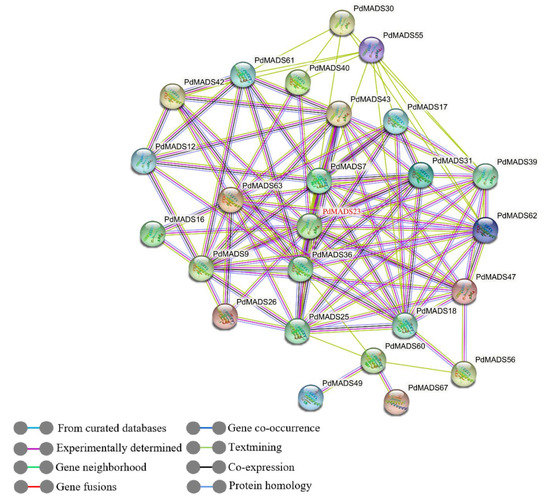
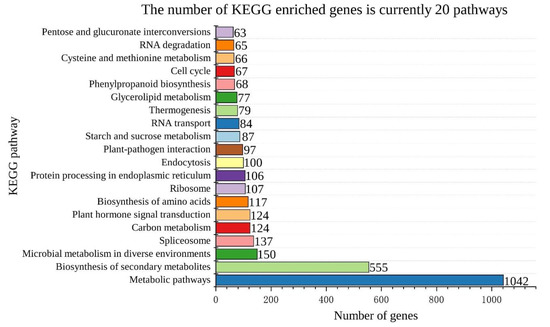
Background: The WRKY transcription factor family represents one of the most crucial transcription factor families in plants, regulating diverse physiological processes. The heartwood of Dalbergia odorifera is a prized material for both high-quality rosewood and traditional medicinal applications, exhibiting exceptional economic value. However, the roles of WRKY transcription factors in the growth and development of D. odorifera, particularly their correlation with heartwood formation, remain unexplored. Methods: WRKY transcription factors were identified through bioinformatics analysis using the published genome data of D. odorifera. Phylogenetic comparative analysis was performed based on the Arabidopsis classification system. Collinearity analysis was conducted to investigate the evolutionary dynamics and expansion mechanisms of the WRKY gene family, and differential expression analysis was performed across tissues. Results: A total of 94 WRKY genes were unevenly distributed across 10 chromosomes and systematically designated as DodWRKY1 to DodWRKY94 according to their chromosomal positions. The WRKY family was classified into three major clades (Groups I, II, and III), with Group II further subdivided into five subgroups (IIa–IIe). Purifying selection served as the primary force shaping the WRKY family, with whole-genome or segmental duplication acting as the dominant expansion mechanism; these duplication events contributed to functional divergence, whereas genes within the same subgroup retained conserved structural features and motif compositions. DodWRKY14 (subgroup IIb) and DodWRKY58/68 (subgroup IIc) were highly expressed in the transition zone, suggesting a potential involvement in heartwood formation. Conclusions: This study provides a comprehensive characterization of the DodWRKY family and identifies candidate genes associated with heartwood formation, thereby establishing a foundation for further investigation into the molecular mechanisms underlying heartwood development.
Full article

Figure 1















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}