




Int. J. Mol. Sci. 2026, 27(8), 3533; https://doi.org/10.3390/ijms27083533 - 15 Apr 2026
Viewed by 530
Abstract
►
Show Figures
Accurate identification of disease-associated microRNAs (miRNAs) is crucial for elucidating pathogenic mechanisms and advancing therapeutic discovery. Although computational methods, particularly those based on biological networks, have become essential tools for predicting miRNA-disease associations, existing approaches often struggle to comprehensively learn from heterogeneous data
[...] Read more.
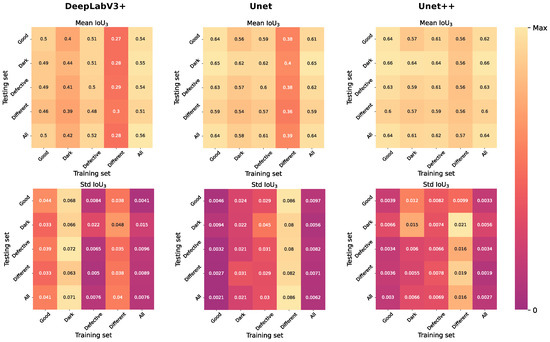
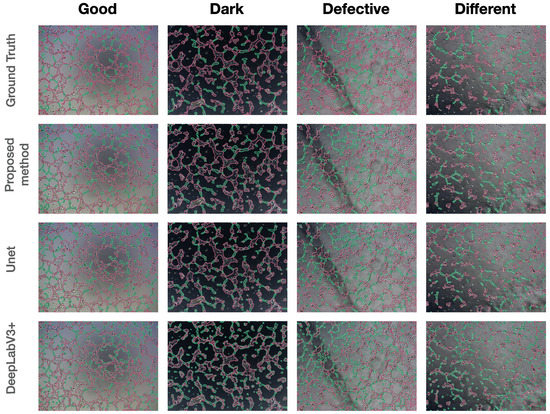
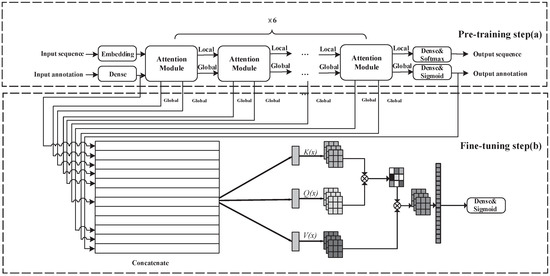
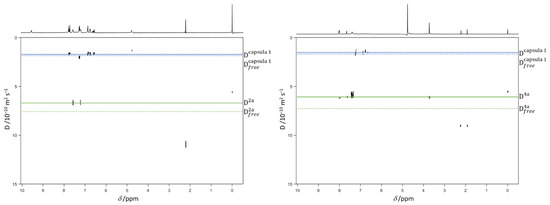
Accurate identification of disease-associated microRNAs (miRNAs) is crucial for elucidating pathogenic mechanisms and advancing therapeutic discovery. Although computational methods, particularly those based on biological networks, have become essential tools for predicting miRNA-disease associations, existing approaches often struggle to comprehensively learn from heterogeneous data and optimize feature representations. To overcome these limitations, we propose the Multi-view Hybrid Attention Graph Convolutional Network (MV-HAGCN). This framework constructs a comprehensive heterogeneous network by integrating multi-source biological information, simultaneously capturing miRNA similarity and disease similarity. We design a hierarchical attention mechanism to enable refined feature learning: first, the Efficient Channel Attention (ECA) module prioritizes information-rich input features, ensuring the model focuses on high-value biological characteristics. Subsequently, the Multi-Head Self-Attention Graph Convolutional Network operates on these refined features. Through iterative message passing and multi-head self-attention, it captures not only direct first-order relationships between nodes but also explicitly models and infers complex, indirect higher-order relationships within the network. This hierarchical design progressively refines feature representations, from channel-level recalibration to global structural dependency modeling, enabling the model to capture both local and high-order relational patterns. Furthermore, a dynamic weight learning strategy adaptively integrates multi-perspective similarity matrices, achieving superior feature complementarity and synergy. Finally, the high-order node representations learned through multi-layer graph convolutions are fed into a multi-layer perceptron for integration and nonlinear transformation, enabling precise prediction of potential miRNA-disease associations. Comprehensive evaluation through five-fold cross-validation on HMDD v2.0 and v3.2 benchmark datasets demonstrates that MV-HAGCN consistently outperforms existing state-of-the-art methods in predictive performance. Case studies targeting key diseases such as breast cancer, lung tumors, and pancreatic disorders revealed that the top 50 miRNAs associated with each of these three conditions were all validated in databases, confirming the practical value of this model in screening candidate miRNAs with high biological relevance.
Full article

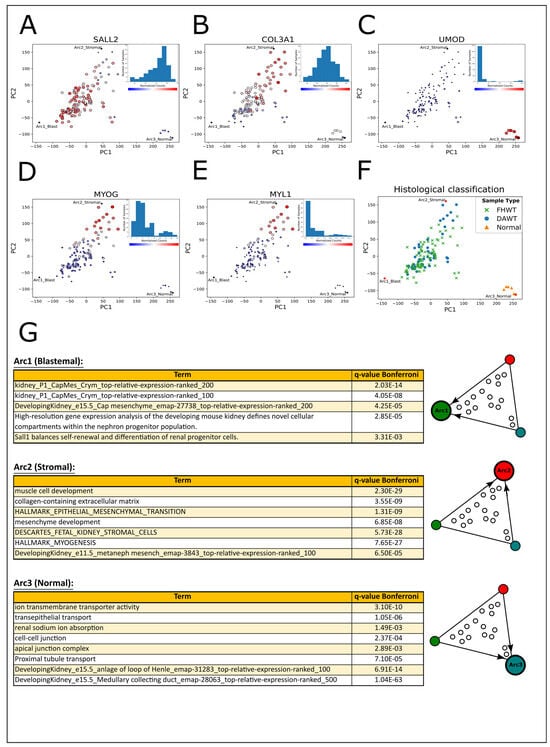
Figure 1

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}