


Thalass. Rep. 2026, 16(2), 9; https://doi.org/10.3390/thalassrep16020009 (registering DOI) - 9 May 2026
Abstract
Background: Advances in medical genetics and prenatal diagnosis have improved the detection of fetal abnormalities during pregnancy. The findings may lead some couples to consider termination of pregnancy (TOP). This study aimed to explore parental perspectives on prenatal diagnosis and termination of pregnancy
[...] Read more.
Background: Advances in medical genetics and prenatal diagnosis have improved the detection of fetal abnormalities during pregnancy. The findings may lead some couples to consider termination of pregnancy (TOP). This study aimed to explore parental perspectives on prenatal diagnosis and termination of pregnancy among families in which both parents were β-thalassemia carriers and had at least one previously affected child. Methods: A qualitative study was conducted using semi-structured interviews with 30 participants (15 fathers and 15 mothers) recruited from Bahawal Victoria Hospital, Bahawalpur, Pakistan, between November 2024 and February 2025. Eligible couples were registered for chorionic villus sampling (CVS)-based prenatal diagnosis; both parents had confirmed β-thalassemia carrier status, and each family had at least one previously affected child with β-thalassemia major or intermedia. Interview data were analyzed using thematic analysis. Results: Religious beliefs, financial burden, prior experience with affected children, and partner support emerged as major influences on reproductive decision-making. Many parents viewed prenatal diagnosis as important for preparation and informed decision-making. Mothers more often described emotional conflict, stress, and reliance on support, whereas some fathers expressed greater acceptance of termination in the context of severe disease burden. Conclusions: Religious beliefs, prior disease experience, family dynamics, and socioeconomic pressures were important and interrelated influences on decisions about prenatal diagnosis and termination within this study population. Our findings underscore the importance of culturally sensitive, non-directive genetic counseling in low-resource settings. The study was limited by its small sample, single-center design, the use of joint spousal interviews, and the possibility that pre-interview counseling influenced participants’ responses.
Full article
(This article belongs to the Section Conventional Treatment of Thalassemia)
►
Show Figures
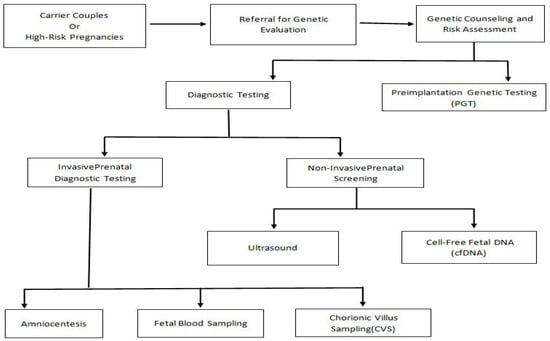
Figure 1

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}