- Review

Diverse Forms of Autophagy and Their Roles in Liver Disease and Aging: A Comprehensive Review
- Seoyoon Heo,
- Min Young Lee and
- Ji Hye Jun
- + 2 authors
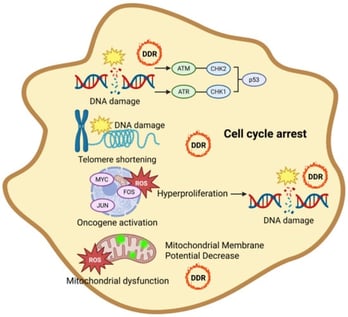
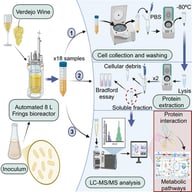
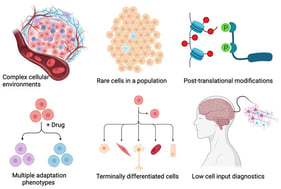
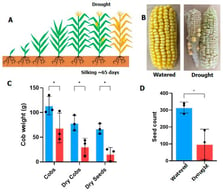
The liver is a central metabolic organ that integrates nutrient sensing, lipid handling, and detoxification to maintain systemic homeostasis. In metabolic dysfunction–associated steatotic liver disease (MASLD), chronic metabolic overload accelerates hepatocyte senescence, impairing regenerative capacity and promoting progression toward fibrosis and hepatocellular carcinoma. While transcriptomic studies have provided important insights into stress-responsive pathways, they incompletely capture the proteome remodeling and proteoform-level alterations that govern hepatocyte function during aging and disease. Recent mass spectrometry–based proteomics studies have revealed that disruption of autophagy-dependent proteome homeostasis is a defining feature of senescent hepatocytes. Quantitative analyses demonstrate coordinated alterations in selective autophagy pathways—including lipophagy, mitophagy, ferritinophagy, ER-phagy, and pexophagy—accompanied by organelle-specific protein abundance signatures and remodeling of autophagy-related proteoforms. These findings position proteomics as an essential tool for resolving the spatial and functional reorganization of hepatocyte proteomes that cannot be inferred from transcript abundance alone. In this review, we synthesize proteomics-driven evidence defining selective autophagy dysfunction in aging and MASLD livers, critically evaluate methodological limitations, and propose a conceptual framework in which impaired selective autophagy acts as a proteome-level driver of hepatocyte senescence. We further outline future directions for proteoform-resolved and spatial proteomics approaches aimed at identifying actionable targets for therapeutic intervention in liver disease.
27 May 2026