
SDS Depletion from Intact Membrane Proteins by KCl Precipitation Ahead of Mass Spectrometry Analysis


Abstract

1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Total Proteome Extraction
2.3. Membrane Proteome Preparations
2.4. KDS Precipitation
2.5. SDS Quantification
2.6. Peptide/Protein Recovery Determination
2.7. SDS-PAGE
2.8. Protein Digestion
2.9. LC-MS Analysis
2.10. Data Analysis
3. Results
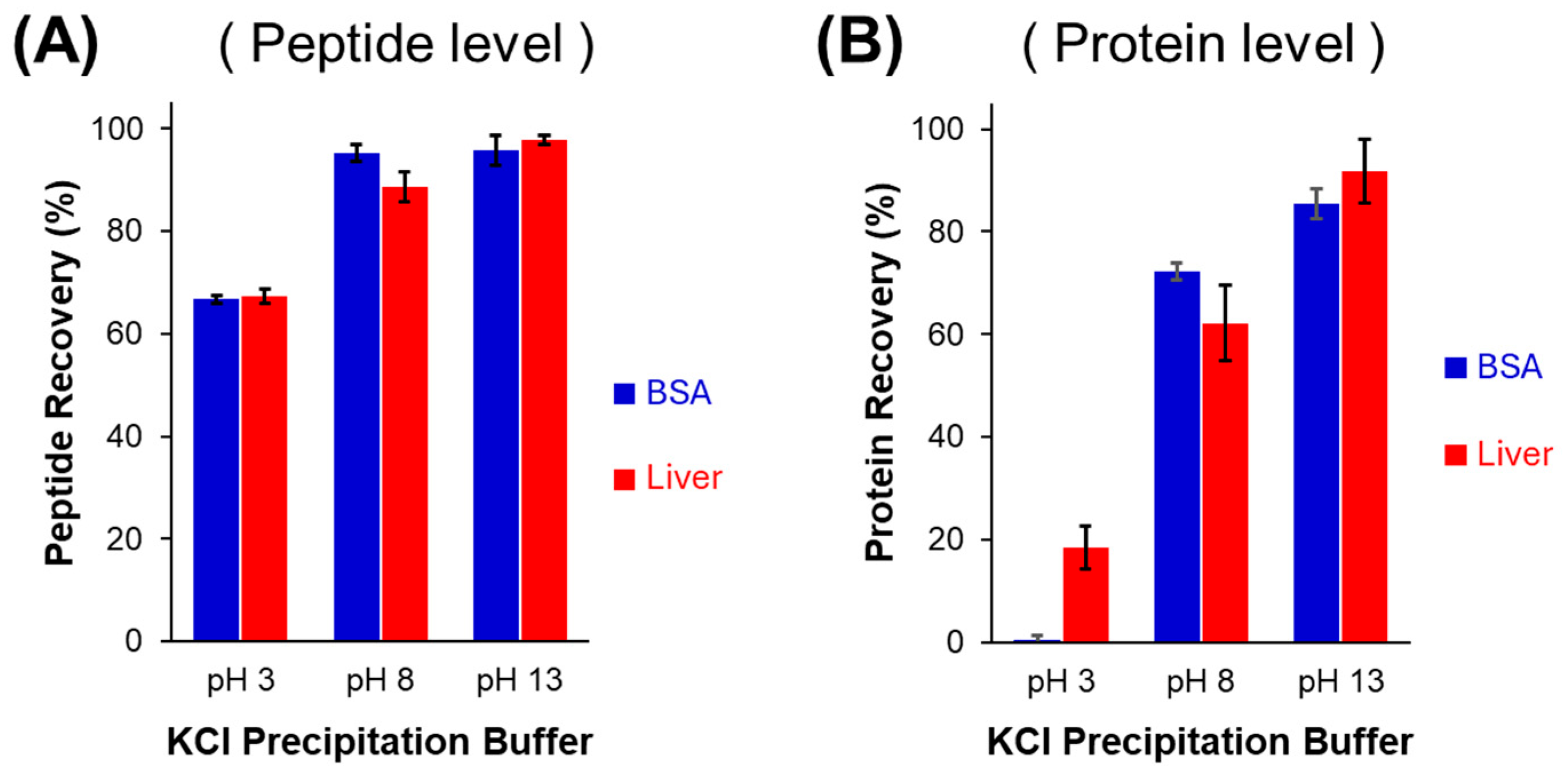
3.1. Peptide Level vs. Protein Level SDS Purification
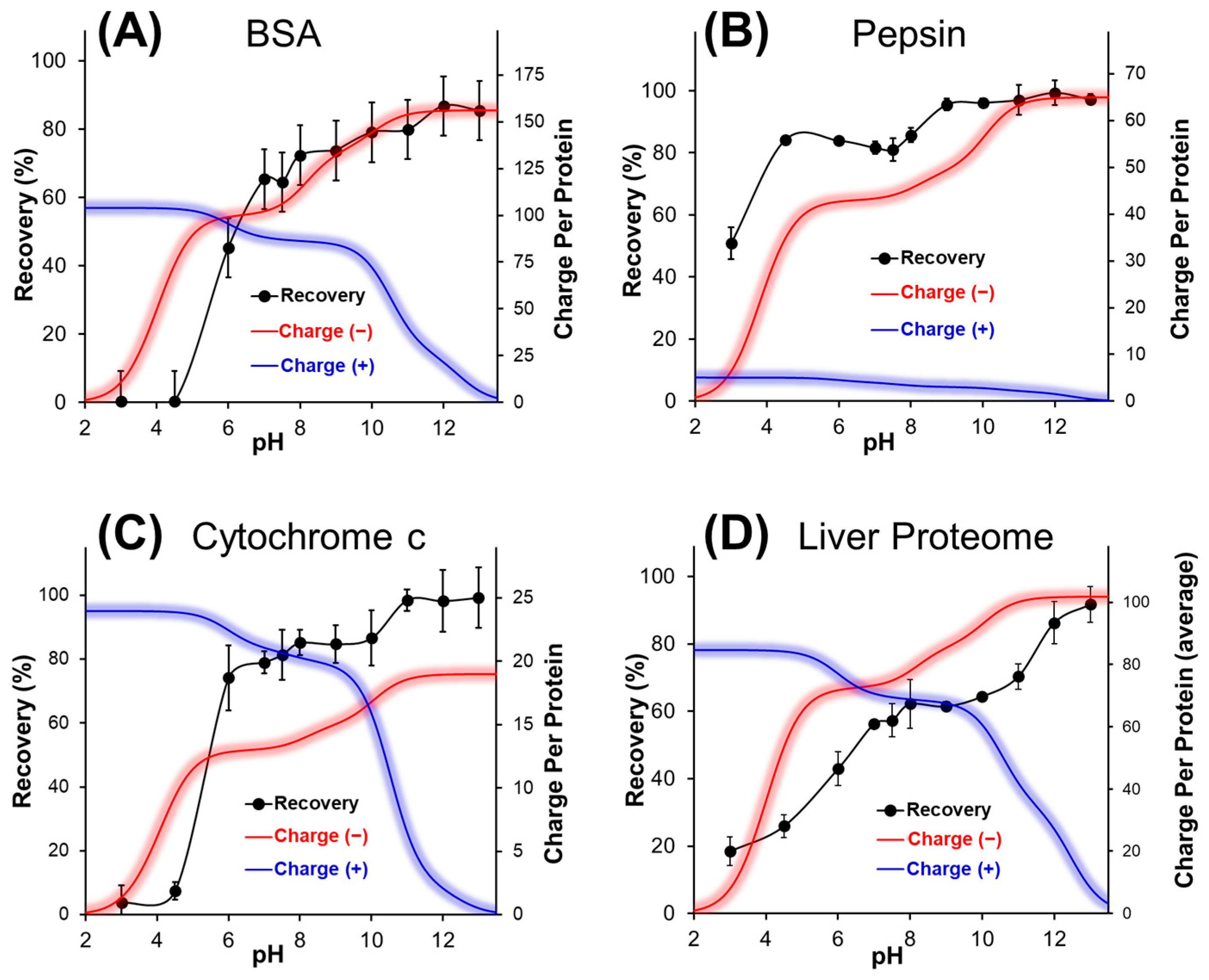
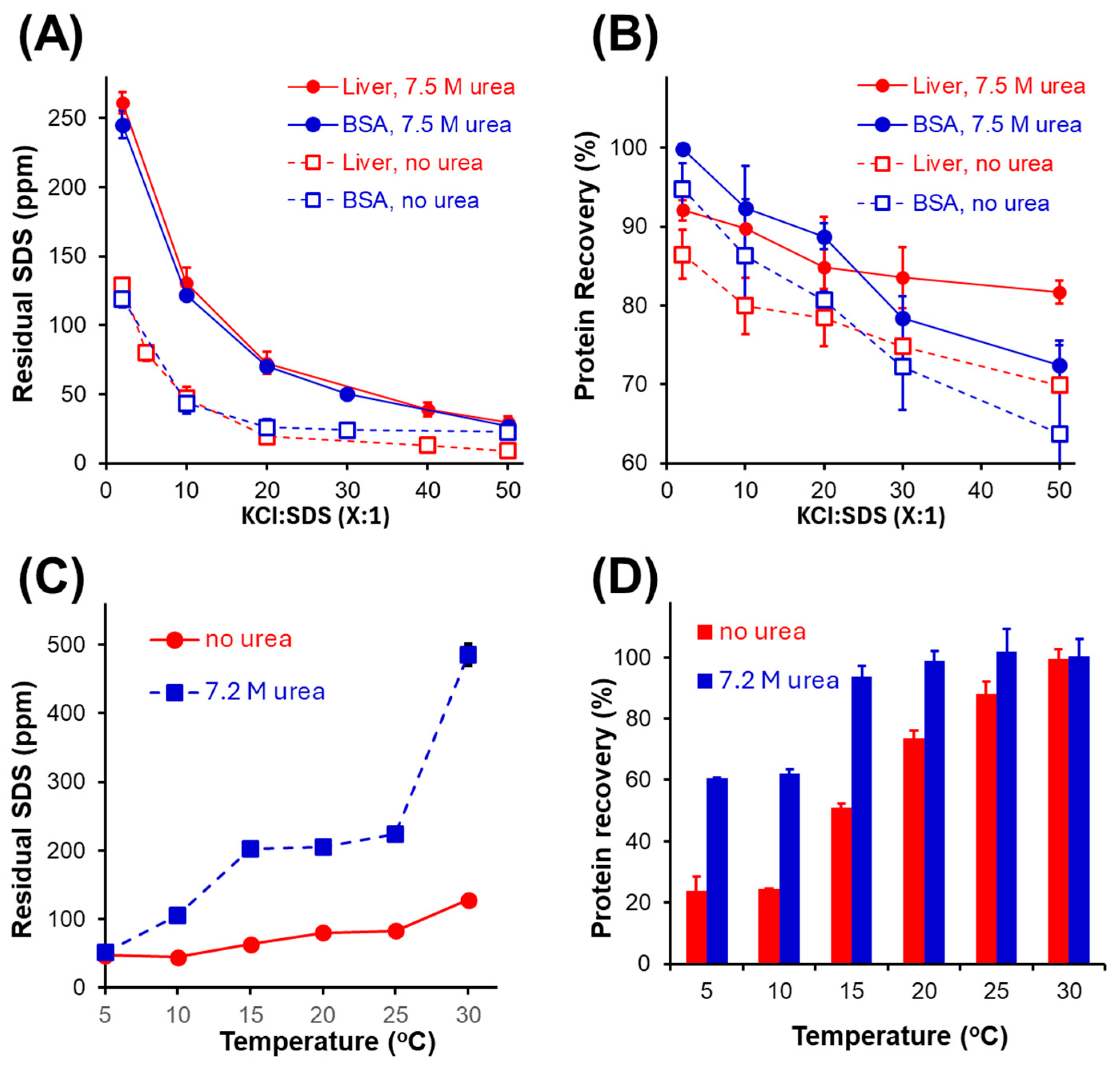
3.2. Variables Impacting Protein Recovery and SDS Precipitation Efficiency
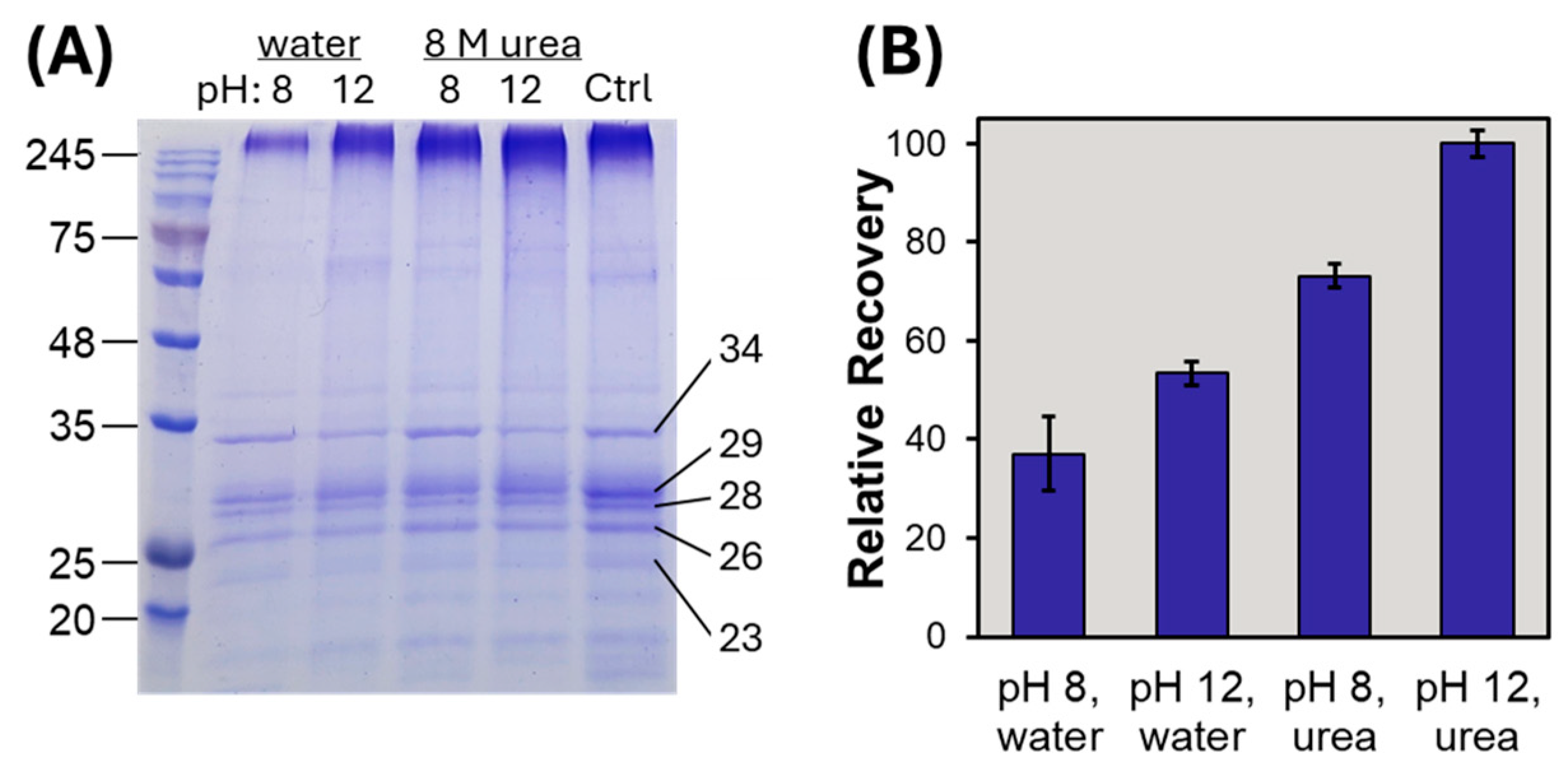
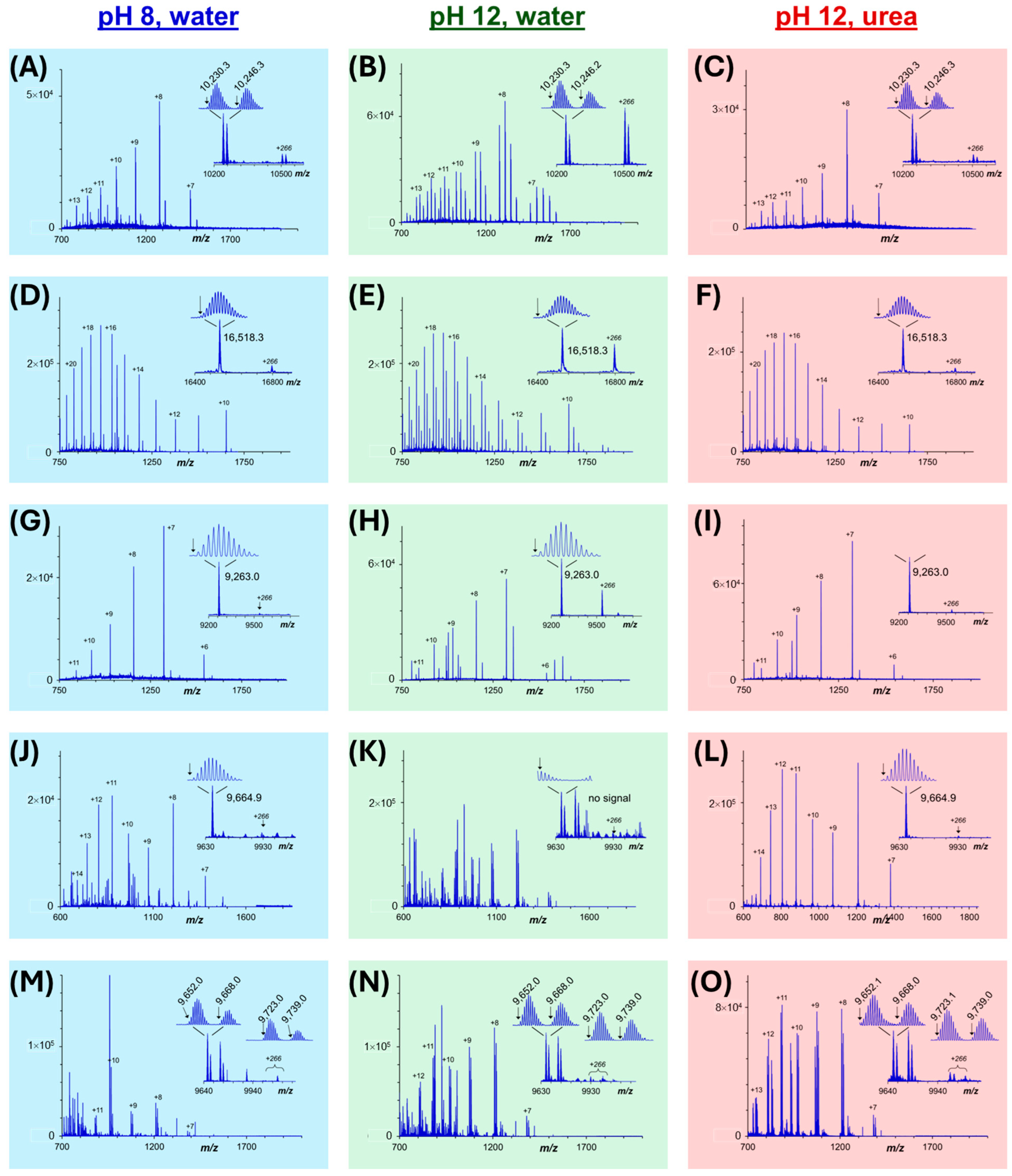
3.3. Intact Membrane Proteome Analysis by Mass Spectrometry After SDS Removal
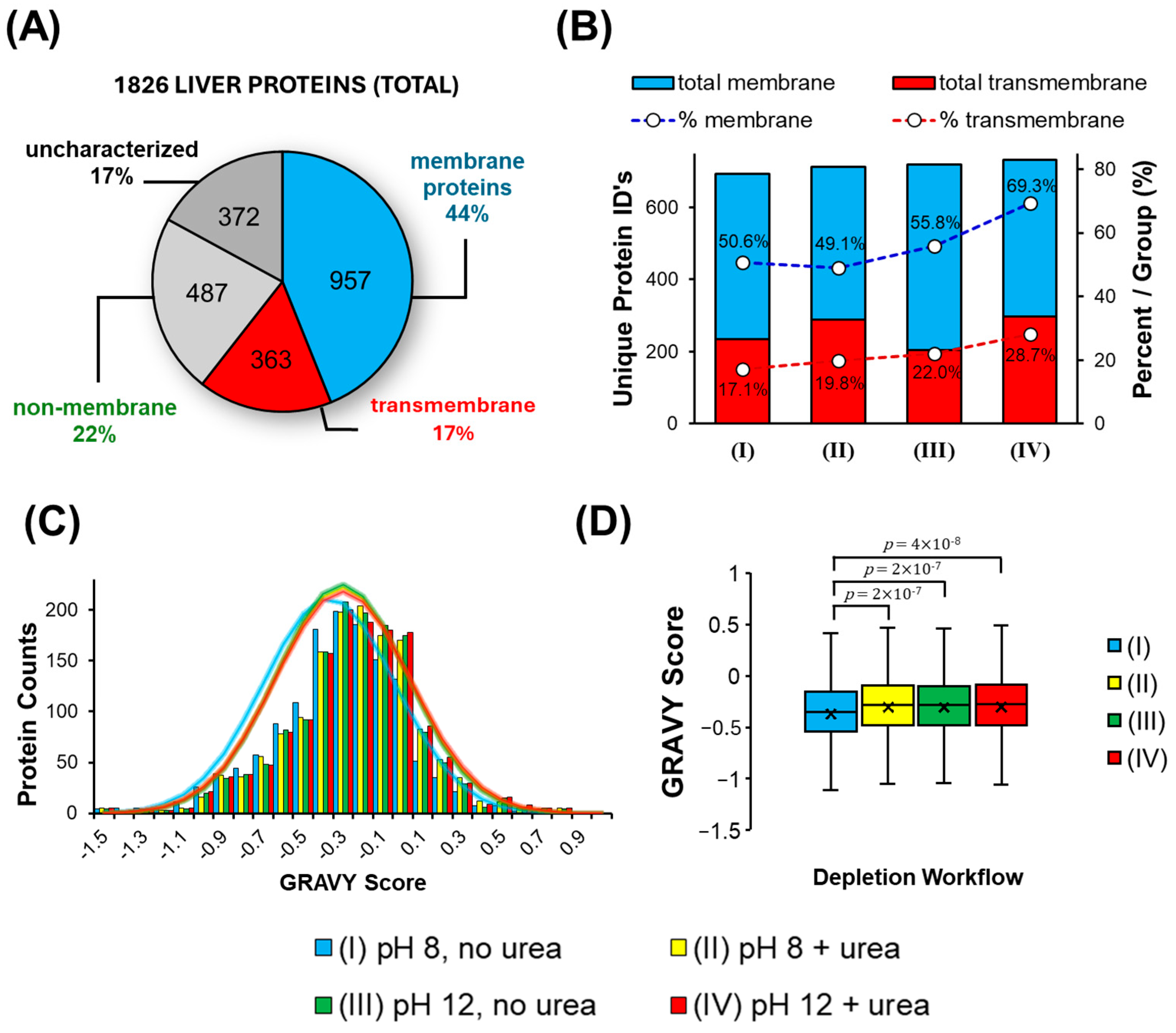
3.4. Bottom-Up Membrane Proteome Analysis Following SDS Precipitation
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| BCA | Bicinchoninic acid |
| BSA | Bovine serum albumin |
| CID | Collisional-induced dissociation |
| DoE | Design of experiments |
| DTT | Dithiothreitol |
| FASP | Filter-aided sample preparation |
| FWHM | Full width at half maximum |
| IDs | Identifications |
| KDS | Potassium dodecyl sulfate |
| GRAVY | Grand average of hydropathy index |
| HPLC | High-performance liquid chromatography |
| LC-UV | Liquid chromatography—ultraviolet absorbance |
| LC-MS | Liquid chromatography—mass spectrometry |
| MBAS | Methylene blue active substances |
| m/z | Mass to charge ratio |
| S/N | Signal-to-noise |
| SDS | Sodium dodecyl sulfate |
| sp3 | Single-pot, solid-phase-enhanced sample preparation |
| STrap | Suspension trapping |
| RF | Radio frequency |
| TFA | Trifluoroacetic acid |
| TPCK | Tosyl phenylalanyl chloromethyl ketone |
References
- Masuda, T.; Tomita, M.; Ishihama, Y. Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 2008, 7, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Feist, P.; Hummon, A.B. Proteomic challenges: Sample preparation techniques for Microgram-Quantity protein analysis from biological samples. Int. J. Mol. Sci. 2015, 16, 3537–3563. [Google Scholar] [CrossRef]
- Botelho, D.; Wall, M.J.; Vieira, D.B.; Fitzsimmons, S.; Liu, F.; Doucette, A. Top-down and bottom-up proteomics of SDS-containing solutions following mass-based separation. J. Proteome Res. 2010, 9, 2863–2870. [Google Scholar] [CrossRef]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.S.; Moggridge, S.; Müller, T.; Sorensen, P.H.; Morin, G.B.; Krijgsveld, J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat. Protoc. 2019, 14, 68–85. [Google Scholar] [CrossRef]
- Crowell, A.M.J.; Wall, M.J.; Doucette, A.A. Maximizing recovery of water-soluble proteins through acetone precipitation. Anal. Chim. Acta 2013, 796, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Nickerson, J.L.; Doucette, A.A. Rapid and quantitative protein precipitation for proteome analysis by mass sectrometry. J. Proteome Res. 2020, 19, 2035–2042. [Google Scholar] [CrossRef]
- Donnelly, D.P.; Rawlins, C.M.; DeHart, C.J.; Fornelli, L.; Schachner, L.F.; Lin, Z.; Lippens, J.L.; Aluri, K.C.; Sarin, R.; Chen, B.; et al. Best practices and benchmarks for intact protein analysis for top-down mass spectrometry. Nat. Methods 2019, 16, 587–594. [Google Scholar] [CrossRef]
- Ilavenil, S.; Al-Dhabi, N.A.; Srigopalram, S.; Kim, Y.O.; Agastian, P.; Baaru, R.; Choi, K.C.; Arasu, M.V.; Park, C.G.; Park, K.H. Removal of SDS from biological protein digests for proteomic analysis by mass spectrometry. Proteome Sci. 2016, 14, 11. [Google Scholar] [CrossRef]
- Erde, J.; Loo, R.R.O.; Loo, J.A. Enhanced FASP (eFASP) to increase proteome coverage and sample recovery for quantitative proteomic experiments. J. Proteome Res. 2014, 13, 1885–1895. [Google Scholar] [CrossRef]
- Liu, Y.; Lin, Y.; Yan, Y.; Li, J.; He, Q.; Chen, P.; Wang, X.; Liang, S. Electrophoretically driven SDS removal and protein fractionation in the shotgun analysis of membrane proteomes. Electrophoresis 2012, 33, 316–324. [Google Scholar] [CrossRef]
- Sun, D.; Wang, N.; Li, L. Integrated SDS removal and peptide separation by strong-cation exchange liquid chromatography for SDS-assisted shotgun proteome analysis. J. Proteome Res. 2012, 11, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rusnak, F. Removal of sodium dodecyl sulfate from protein and peptide samples with cross-linked [Os(dmebpy)2Cl]+/2+-derivatized acrylamide and vinylimidazole copolymer. Rapid Commun. Mass Spectrom. 2016, 30, 698–704. [Google Scholar] [CrossRef]
- Kachuk, C.; Faulkner, M.; Liu, F.; Doucette, A.A. Automated SDS depletion for mass spectrometry of intact membrane proteins though transmembrane electrophoresis. J. Proteome Res. 2016, 15, 2634–2642. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.M.; Floyd, E.; Baker, E.S.; Zhao, R.; Wu, S.; Paša-Tolić, L. Evaluation of SDS depletion using an affinity spin column and IMS-MS detection. Proteomics 2012, 12, 3138–3142. [Google Scholar] [CrossRef] [PubMed]
- Carraro, U.; Doria, D.; Rizzi, C.; Sandri, M. A new two-step precipitation method removes free-SDS and thiol reagents from diluted solutions, and then allows recovery and quantitation of proteins. Biochem. Biophys. Res. Commun. 1994, 200, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Van Heyningen, S. The Effect of K+ and pH on Protein-Dodecylsulphate Complexes. Biochim. Biophys. Acta—Protein Struct. 1973, 328, 303–313. [Google Scholar] [CrossRef]
- Carraro, U.; Rizzi, C.; Sandri, M. Effective recovery by KCl precipitation of highly diluted muscle proteins solubilized with sodium dodecyl sulfate. Electrophoresis 1991, 12, 1005–1010. [Google Scholar] [CrossRef]
- Zaidi, N.; Nusrat, S.; Zaidi, F.K.; Khan, R.H. pH-dependent differential interacting mechanisms of sodium dodecyl sulfate with bovine serum fetuin: A biophysical insight. J. Phys. Chem. B 2014, 118, 13025–13036. [Google Scholar] [CrossRef]
- Zhou, J.-Y.; Dann, G.P.; Shi, T.; Wang, L.; Gao, X.; Su, D.; Nicora, C.D.; Shukla, A.K.; Moore, R.J.; Liu, T.; et al. Simple Sodium Dodecyl Sulfate-Assisted Sample Preparation Method for LC-MS-Based Proteomics Applications. Anal. Chem. 2012, 84, 2862–2867. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M.; Rizzi, C.; Catani, C.; Carraro, U. Selective removal of free dodecyl sulfate from 2-mercaptoethanol-SDS-solubilized proteins before KDS-protein precipitation. Anal. Biochem. 1993, 213, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.K.; Johnson, T.; Yang, Y.Y.; Shore, V. Selective separation of virus proteins and double-stranded RNAs by SDS-KCl precipitation. J. Virol. Methods 1989, 26, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Winogradoff, D.; John, S.; Aksimentiev, A. Protein unfolding by SDS: The microscopic mechanisms and the properties of the SDS-protein assembly. Nanoscale 2020, 12, 5422–5434. [Google Scholar] [CrossRef]
- Tummino, P.J.; Gafni, A. Determination of the aggregation number of detergent micelles using steady-state fluorescence quenching. Biophys. J. 1993, 64, 1580–1587. [Google Scholar] [CrossRef]
- Takeda, K.; Wada, A.; Sato, Y.; Hamada, S. Removal of dodecyl sulfate ions bound to bovine serum albumin and chymotrypsinogen from the proteins. Effects of reduction of disulfide bridges and cleavage of peptide bonds on the removal extent. J. Colloid Interface Sci. 1991, 147, 51–56. [Google Scholar] [CrossRef]
- Žilionis, A. Removal of sodium dodecyl sulfate from protein samples. Chemija 2018, 29, 199–208. [Google Scholar] [CrossRef]
- Fujiki, Y.; Hubbard, A.L.; Fowler, S.; Lazarow, P.B. Isolation of intracellular membranes by means of sodium carbonate treatment: Application to endoplasmic reticulum. J. Cell Biol. 1982, 93, 97–102. [Google Scholar] [CrossRef]
- Arand, M.; Friedberg, T.; Oesch, F. Colorimetric quantitation of trace amounts of sodium lauryl sulfate in the presence of nucleic acids and proteins. Anal. Biochem. 1992, 207, 73–75. [Google Scholar] [CrossRef]
- Orton, D.J.; Doucette, A.A. A universal, high recovery assay for protein quantitation through temperature programmed liquid chromatography (TPLC). J. Chromatogr. B 2013, 921–922, 75–80. [Google Scholar] [CrossRef]
- Harris, D.C.; Lucy, C.A. Quantitative Chemical Analysis, 10th ed.; W.H. Freeman & Company: New York, NY, USA, 2020. [Google Scholar]
- Espinosa, Y.R.; Grigera, R.J.; Ferrara, C.G. Mechanisms associated with the effects of urea on the micellar structure of sodium dodecyl sulphate in aqueous solutions. Prog. Biophys. Mol. Biol. 2018, 140, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Grefrath, S.P.; Reynolds, J.A. The molecular weight of the major glycoprotein from the human erythrocyte membrane. Proc. Natl. Acad. Sci. USA 1974, 71, 3913–3916. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strategy | Description | Applies to Protein? | Applies to Peptide? | Potential Concerns | Refs |
|---|---|---|---|---|---|
| Solvent precipitation | Pellet the proteins in organic solvent (e.g., acetone, chloroform/methanol), while the detergent remains in solution; decant. | Yes | No | Proteins must be resolubilized, lowering throughput and potentially introducing sample loss. | [7] |
| Filter-Aided Sample Preparation (FASP) | Retain intact proteins on molecular weight filter, adding urea to weaken SDS–protein interactions; digest proteins on filter, and elute as peptides. | Yes | No | Variable protein recovery from binding to the molecular weight filters. Slow, labor-intensive processing. | [4] |
| Suspension Trapping (Strap) | Acid + methanol-precipitated proteins are retained on glass filter cartridge, digested, and released as peptides. | Yes | No | Commercial cartridges (higher cost). | [5] |
| Single-pot, solid-phase-enhanced sample preparation (sp3) | Capture intact proteins in high organic solvent on hydrophilic interaction magnetic beads, add enzyme to digest on-bead, elute the digestion products | Yes | No | Commercial cartridges (higher cost). | [6] |
| Metal–Organic Frameworks (MOF) | SDS retention on metal-containing nanoporous resin. | Yes | Yes | Non-selective interactions; little quantitative data on SDS removal efficiency. | [10] |
| Enhanced Filter-Aided Sample Preparation (eFASP) | Substitute the urea in the conventional FASP approach with sodium deoxycholate. | Yes | No | Lengthy, labor-intensive processing. | [11] |
| Tube-Gel Electrophoresis | Load samples into SDS PAGE gel tubes. Conventional in-gel digestion. Extensive washing releases the surfactant; trypsin releases the peptides. | Yes | No | Lengthy processing, and risk of sample loss within the gel. | [12] |
| Strong Cation Exchange (SCX) | Inject peptides + SDS onto cation exchange column, retaining positive peptides in acid and washing away negatively charged SDS. | No | Yes | SDS–protein interactions suggest surfactant retention on column, leading to low purity. | [13] |
| KCl | Potassium dodecyl sulfate has minimal solubility in water, thus precipitating the SDS while peptides remain in solution. | Yes (*) | Yes | SDS–protein interactions can lead to co-precipitation of proteins. | [14] |
| Transmembrane Electrophoresis (TME) | Protein dialysis in an electric field which forces free and protein-bound SDS to migrate through the dialysis membrane, retaining intact proteins in solution. | Yes | No | Custom device which must be manufactured for use. | [15] |
| PierceTM Detergent Removal Spin Cartridge (Thermo Fisher Scientific, Waltham, MA, USA) | Proprietary resin in spin-column format which captures SDS, allowing peptides to pass through the resin. | Yes | Yes | Commercial cartridges (higher cost). | [16] |
| ProteoSpinTM Detergent Cleanup Kit (Thermo Fisher Scientific) | Proprietary resin in spin-column format which captures SDS, allowing peptides to pass through the resin. | Yes | No | Commercial kit (higher cost). | - |
| SDS-OutTM SDS precipitation kit (Thermo Fisher Scientific) | Reagent kit to induce precipitation of SDS while retaining proteins in solution | Yes | Yes | Commercial kit (higher cost). | - |
| Protein Recovery | SDS Depletion | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Variable | Coefficient | Lower 95% | Upper 95% | t-Statistic | p-Value | Coefficient | Lower 95% | Upper 95% | t-Statistic | p-Value |
| (A) pH | −0.0053 | −0.03 | 0.02 | −0.5 | 0.62 | 21.5 | 19.5 | 23.5 | 23.28 | 1.4 × 10−12 |
| (B) KCl | −0.4 | −0.43 | −0.38 | −38.3 | 2.3 × 10−16 | −2.4 | −4.3 | −0.4 | −2.55 | 0.023 |
| (C) urea | 0.115 | 0.09 | 0.14 | 10.9 | 1.6 × 10−8 | 19.3 | 17.3 | 21.3 | 20.9 | 5.9 × 10−12 |
| AB | 0.022 | 0 | 0.04 | 2.1 | 0.053 | −2.1 | −4.1 | −0.2 | −2.31 | 0.036 |
| AC | −0.028 | −0.05 | −0.01 | −2.65 | 0.018 | 17.9 | 15.9 | 19.8 | 19.36 | 1.7 × 10−11 |
| BC | 0.036 | 0.01 | 0.06 | 3.41 | 3.9 × 10−3 | −1.7 | −3.6 | 0.3 | −1.8 | 0.093 |
| ABC | 0.035 | 0.01 | 0.06 | 3.26 | 5.2 × 10−3 | −1.6 | −3.6 | 0.4 | −1.7 | 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iranpour, T.; Mirimba, M.; Shenouda, C.; Lynch, A.; Doucette, A.A. SDS Depletion from Intact Membrane Proteins by KCl Precipitation Ahead of Mass Spectrometry Analysis. Proteomes 2025, 13, 30. https://doi.org/10.3390/proteomes13030030
Iranpour T, Mirimba M, Shenouda C, Lynch A, Doucette AA. SDS Depletion from Intact Membrane Proteins by KCl Precipitation Ahead of Mass Spectrometry Analysis. Proteomes. 2025; 13(3):30. https://doi.org/10.3390/proteomes13030030
Chicago/Turabian StyleIranpour, Tania, Mapenzi Mirimba, Chloe Shenouda, Adam Lynch, and Alan A. Doucette. 2025. "SDS Depletion from Intact Membrane Proteins by KCl Precipitation Ahead of Mass Spectrometry Analysis" Proteomes 13, no. 3: 30. https://doi.org/10.3390/proteomes13030030
APA StyleIranpour, T., Mirimba, M., Shenouda, C., Lynch, A., & Doucette, A. A. (2025). SDS Depletion from Intact Membrane Proteins by KCl Precipitation Ahead of Mass Spectrometry Analysis. Proteomes, 13(3), 30. https://doi.org/10.3390/proteomes13030030