


Genes 2025, 16(9), 1061; https://doi.org/10.3390/genes16091061 - 9 Sep 2025
Abstract
Background/Objectives: Twin and family studies suggest that 90% of the risk for autism spectrum disorder (ASD) is due to genetic factors, with 800 genes recognized as playing a role. An important gene is phosphatase and tensin homolog (PTEN), which plays
[...] Read more.
Background/Objectives: Twin and family studies suggest that 90% of the risk for autism spectrum disorder (ASD) is due to genetic factors, with 800 genes recognized as playing a role. An important gene is phosphatase and tensin homolog (PTEN), which plays a significant role in cancer as a tumor suppressor best known for causing overgrowth and PTEN hamartoma tumor syndromes (PHTS). Less well known are PTEN germline mutations with adverse neurodevelopmental impacts of macrocephaly, intellectual disability, and ASD, as well as other behavioral and psychiatric disturbances. There remains a limited understanding of whether these gene variants are associated with differing manifestations of PTEN-associated neurodevelopmental disorders. Methods: This review utilized comprehensive literature searches such as PubMed, OMIM, and Gene Reviews with keywords of PTEN, genetic factors, autism, and human studies and by searching genomic-protein functional networks with STRING computer-based programs for functional and genetic mechanisms. Results: This review explored the genetic underpinnings of PTEN gene variants causing altered interactive proteins and their mechanisms, biological processes, molecular functions, pathways, and disease–gene associations. We characterized specific gene–gene or protein–protein interactions and their functions relating to neurodevelopment, psychiatric disorders, and ASD that were found to be increased with PTEN gene variants. Conclusions: PTEN gene defects are among the most recognized genetic causes of ASD. PTEN gene variants and altered protein interactions and mechanisms described in our study are associated with an increased risk for tissue and organ overgrowth, macrocephaly, and distinct brain anomalies, specifically newly identified abnormal CSF dynamics. These genetic underpinnings and impacts on neurodevelopment are discussed. The genetic and protein findings identified may offer clues to effective treatment interventions, particularly when instituted at a young age, to improve long-term outcomes.
Full article
(This article belongs to the Section Neurogenomics)
►
Show Figures

Figure 1




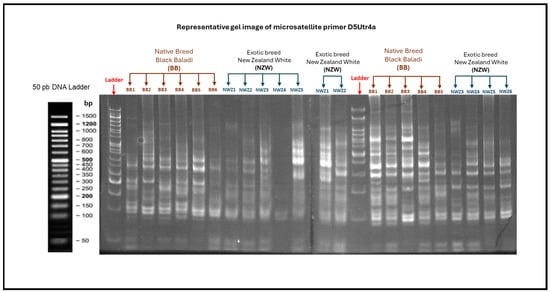










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}