



Cardiogenetics 2026, 16(2), 12; https://doi.org/10.3390/cardiogenetics16020012 - 3 Jun 2026
Abstract
Background: Heterozygous familial hypercholesterolemia (HeFH) is characterized by elevated low-density lipoprotein cholesterol (LDL-C) and increased cardiovascular risk, making early diagnosis essential; however, the diagnostic performance of pediatric criteria is heterogeneous. This study evaluated the effectiveness of different diagnostic criteria and scoring systems to
[...] Read more.
Background: Heterozygous familial hypercholesterolemia (HeFH) is characterized by elevated low-density lipoprotein cholesterol (LDL-C) and increased cardiovascular risk, making early diagnosis essential; however, the diagnostic performance of pediatric criteria is heterogeneous. This study evaluated the effectiveness of different diagnostic criteria and scoring systems to select children for genetic testing. Materials and methods: A total of 214 pediatric subjects with suspected HeFH were included, recruited from patients followed at a tertiary care center, based on LDL-C levels ≥ 95th age- and sex-specific percentile in both the proband and one biological parent. All subjects underwent genetic analysis of the main FH-associated genes (LDLR, APOB, PCSK9). The following diagnostic criteria and scoring systems were retrospectively evaluated and compared with genetic findings: Simon Broome Register (SBR), Dutch Lipid Clinic Network (DLCN), European Atherosclerosis Society (EAS), American Heart Association (AHA), Familial Hypercholesterolemia Canada Network (FH-CAN), Japanese Atherosclerosis Society (JAS), Lipid TransPort Disorders Italian Genetic Network for Italian pediatric patients (LIPIGEN-FH-PED), and the Familial Hypercholesterolemia Pediatric Diagnostic Score (FH-PeDS). Results: Pathogenic variants were identified in 91.8% of subjects. Approaches using lower LDL-C thresholds minimized the loss of variant-positive individuals (particularly JAS and FH-PeDS, with a missed diagnoses rate of 1.6%), whereas more restrictive definitions excluded a substantial proportion of affected patients (10.5% SBR, 56.3% DLCN, 6.3% EAS, 6.3% AHA, 7.4% FH-CAN, and 6.3% LIPIGEN-FH-PED). The mutation detection rate (MDR) was >91% for all examined criteria. Conclusions: Several current diagnostic criteria may underestimate the true number of children carrying FH-associated variants. Less selective criteria enable the identification of a greater number of FH-positive individuals while maintaining a high MDR, thus supporting the prioritization of identifying as many affected children as possible in the pediatric setting. This cohort reflects a tertiary referral population rather than the general population; therefore, further studies are needed to evaluate the applicability of our findings to broader public health contexts and screening settings.
Full article
(This article belongs to the Section Cardiovascular Genetics in Clinical Practice)
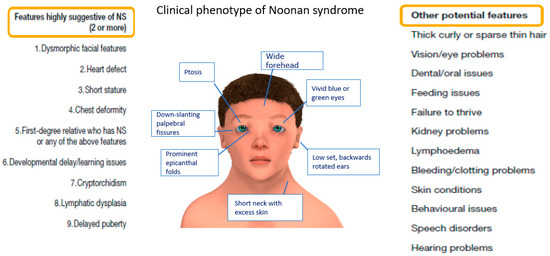






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}