
The Influence of Genotype on the Cardiopulmonary Test Response in Patients Affected by Hypertrophic Cardiomyopathy
 ,
,  , , , , , ,
, , , , , ,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Echocardiography
2.3. Genetic Testing
2.4. Cardiopulmonary Test
2.5. Statistics
3. Results
3.1. Relevance of the Presence of P/LP Mutations
3.2. Relevance of Different Pathogenetic P/LP Mutations
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Makavos, G.; Κairis, C.; Tselegkidi, M.E.; Karamitsos, T.; Rigopoulos, A.G.; Noutsias, M.; Ikonomidis, I. Hypertrophic cardiomyopathy: An updated review on diagnosis, prognosis, and treatment. Heart Fail. Rev. 2019, 24, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Arbelo, E.; Protonotarios, A.; Gimeno, J.R.; Arbustini, E.; Barriales-Villa, R.; Basso, C.; Bezzina, C.R.; Biagini, E.; Blom, N.A.; de Boer, R.A.; et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur. Heart J. 2023, 44, 3503–3626. [Google Scholar] [CrossRef] [PubMed]
- Klues, H.G.; Schiffers, A.; Maron, B.J. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: Morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J. Am. Coll. Cardiol. 1995, 26, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Lopes, L.R.; Rahman, M.S.; Elliott, P.M. A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 2013, 99, 1800–1811. [Google Scholar] [CrossRef]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and Lifetime Burden of Disease in Hypertrophic Cardiomyopathy. Insights From the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Márquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick Eduardo, B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace 2022, 24, 1307–1367. [Google Scholar] [CrossRef]
- Olivotto, I.; Cecchi, F.; Poggesi, C.; Yacoub, M.H. Patterns of disease progression in hypertrophic cardiomyopathy: An individualized approach to clinical staging. Circ. Heart Fail. 2012, 12, 535–546. [Google Scholar] [CrossRef]
- Girolami, F.; Gozzini, A.; Pálinkás, E.D.; Ballerini, A.; Tomberli, A.; Baldini, K.; Marchi, A.; Zampieri, M.; Passantino, S.; Porcedda, G.; et al. Genetic Testing and Counselling in Hypertrophic Cardiomyopathy: Frequently Asked Questions. J. Clin. Med. 2023, 12, 2489. [Google Scholar] [CrossRef]
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat. Genet. 2021, 53, 135–142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease. Cardiac and Vascular Biology; Erdmann, J., Moretti, A., Eds.; Springer: Cham, Switzerland, 2019; Volume 7. [Google Scholar] [CrossRef]
- Maron, B.J.; Maron, M.S.; Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: Clinical perspectives. J. Am. Coll. Cardiol. 2012, 21, 705–715. [Google Scholar] [CrossRef]
- Ommen, S.R.; Ho, C.Y.; Asif, I.M.; Balaji, S.; Burke, M.A.; Day, S.M.; Dearani, J.A.; Epps, K.C.; Evanovich, L.; Ferrari, V.A.; et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2024, 83, 2324–2405. [Google Scholar] [CrossRef] [PubMed]
- Bonaventura, J.; Rowin, E.J.; Chan, R.H.; Chin, M.T.; Puchnerova, V.; Polakova, E.; Macek, M., Jr.; Votypka, P.; Batorsky, R.; Perera, G.; et al. Relationship Between Genotype Status and Clinical Outcome in Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2024, 13, e033565. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Coppini, R.; Ho, C.Y.; Ashley, E.; Day, S.; Ferrantini, C.; Girolami, F.; Tomberli, B.; Bardi, S.; Torricelli, F.; Cecchi, F.; et al. Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J. Am. Coll. Cardiol. 2014, 64, 2589–2600. [Google Scholar] [CrossRef]
- Beltrami, M.; Fedele, E.; Fumagalli, C.; Mazzarotto, F.; Girolami, F.; Ferrantini, C.; Coppini, R.; Tofani, L.; Bertaccini, B.; Poggesi, C.; et al. Long-Term prevalence of systolic dysfunction in MYBPC3 versus MYH7-related hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 2023, 16, 363–371. [Google Scholar] [CrossRef]
- Chumakova, O.S.; Baklanova, T.N.; Zateyshchikov, D.A. Clinical Features and Prospective Outcomes of Thin-Filament Hypertrophic Cardiomyopathy: Intrinsic Data and Comparative Insights from Other Cohorts. J. Clin. Med. 2025, 14, 866. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Finocchiaro, G.; Haddad, F.; Knowles, J.W.; Caleshu, C.; Pavlovic, A.; Homburger, J.; Shmargad, Y.; Sinagra, G.; Magavern, E.; Wong, M.; et al. Cardiopulmonary responses and prognosis in hypertrophic cardiomyopathy: A potential role for comprehensive noninvasive hemodynamic assessment. JACC Heart Fail. 2015, 3, 408–418. [Google Scholar] [CrossRef]
- Magrì, D.; Limongelli, G.; Re, F.; Agostoni, P.; Zachara, E.; Correale, M.; Mastromarino, V.; Santolamazza, C.; Casenghi, M.; Pacileo, G.; et al. Cardiopulmonary exercise test and sudden cardiac death risk in hypertrophic cardiomyopathy. Heart 2016, 102, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Magrì, D.; Santolamazza, C. Cardiopulmonary exercise test in hypertrophic cardiomyopathy. Ann. Am. Thorac. Soc. 2017, 14, S102–S109. [Google Scholar] [CrossRef]
- Magrì, D.; Re, F.; Limongelli, G.; Agostoni, P.; Zachara, E.; Correale, M.; Mastromarino, V.; Santolamazza, C.; Casenghi, M.; Pacileo, G.; et al. Heart Failure Progression in Hypertrophic Cardiomyopathy—Possible Insights From Cardiopulmonary Exercise Testing. Circ. J. 2016, 80, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Magrì, D.; Mastromarino, V.; Gallo, G.; Zachara, E.; Re, F.; Agostoni, P.; Giordano, D.; Rubattu, S.; Forte, M.; Cotugno, M.; et al. Risk Stratification in Hypertrophic Cardiomyopathy. Insights from Genetic Analysis and Cardiopulmonary Exercise Testing. J. Clin. Med. 2020, 9, 1636. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Maron, M.S.; Olivotto, I.; Betocchi, S.; Casey, S.A.; Lesser, J.R.; Losi, M.A.; Cecchi, F.; Maron, B.J. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N. Engl. J. Med. 2003, 348, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Critoph, C.H.; Patel, V.; Mist, B.; Elliott, P.M. Cardiac output response and peripheral oxygen extraction during exercise among symptomatic hypertrophic cardiomyopathy patients with and without left ventricular outflow tract obstruction. Heart 2014, 100, 639–646. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef] [PubMed]
- Nagueh, S.F.; Phelan, D.; Abraham, T.; Armour, A.; Desai, M.Y.; Dragulescu, A.; Gilliland, Y.; Lester, S.J.; Maldonado, Y.; Mohiddin, S.; et al. Recommendations for Multimodality Cardiovascular Imaging of Patients with Hypertrophic Cardiomyopathy: An Update from the American Society of Echocardiography, in Collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J. Am. Soc. Echocardiogr. 2022, 35, 533–569. [Google Scholar] [CrossRef] [PubMed]
- Myers, J.; Arena, R.; Cahalin, L.P.; Labate, V.; Guazzi, M. Cardiopulmonary Exercise Testing in Heart Failure. Curr. Probl. Cardiol. 2015, 40, 322–372. [Google Scholar] [CrossRef] [PubMed]
- Stringer, W.W.; Hansen, J.E.; Wasserman, K. Cardiac output estimated noninvasively from oxygen uptake during exercise. J. Appl. Physiol. 1997, 82, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Zeppenfeld, K.; Tfelt-Hansen, J.; de Riva, M.; Winkel, B.G.; Behr, E.R.; Blom, N.A.; Charron, P.; Corrado, D.; Dagres, N.; de Chillou, C.; et al. 2022 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur. Heart J. 2022, 43, 3997–4126. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Berteotti, M.; Ferrantini, C.; Tassetti, L.; Gabriele, M.; Tomberli, B.; Castelli, G.; Cappelli, F.; Stefàno, P.; Marchionni, N.; et al. Pathophysiology and Treatment of Hypertrophic Cardiomyopathy: New Perspectives. Curr. Heart Fail. Rep. 2021, 18, 169–179. [Google Scholar] [CrossRef]
- Olivotto, I.; Girolami, F.; Sciagrà, R.; Ackerman, M.J.; Sotgia, B.; Bos, J.M.; Nistri, S.; Sgalambro, A.; Grifoni, C.; Torricelli, F.; et al. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. J. Am. Coll. Cardiol. 2011, 58, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Spudich, J.A. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflug. Arch. Eur. J. Physiol. 2019, 471, 701–717. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.; Liu, X.; Sparrow, A.; Patel, S.; Zhang, Y.-H.; Casadei, B.; Watkins, H.; Redwood, C. Hypertrophic cardiomyopathy mutations increase myofilament Ca2+ buffering, alter intracellular Ca2+ handling, and stimulate Ca2+-dependent signaling. J. Biol. Chem. 2018, 293, 10487–10499. [Google Scholar] [CrossRef] [PubMed]
- Keyt, L.K.; Duran, J.M.; Bui, Q.M.; Chen, C.; Miyamoto, M.I.; Enciso, J.S.; Tardiff, J.C.; Adler, E.D. Thin filament cardiomyopathies: A review of genetics, disease mechanisms, and emerging therapeutics. Front. Cardiovasc. Med. 2022, 9, 972301. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Pt Number | Gene | Type Gene | Nucleotide Change | Amino Acid Change | Class | ACMG Criteria |
|---|---|---|---|---|---|---|
| 1 | MYBPC3 | THICK | c.G1624C | p.E542Q | P | PM3, PP1, PS3, PM2, PP3 |
| 2 | MYBPC3 | THICK | c.T3713C | p.L1238P | P | PS4, PP1, PP3, PM2 |
| 3 | MYH7 | THICK | c.G1231A | p.V411I | P | PS4, PM1, PP2, PM2, PP3 |
| 4 | MYBPC3 | THICK | c.1458-1G>A | P | PS4, PVS1, PM2 | |
| 5 | MYH7 | THICK | c.G1063A | p.A355T | P | PP5, PP3, PM1, PM2 |
| 6 | MYH7 | THICK | c.C2080T | p.R694C | LP | PS4, PP1, PM1, PP2, PM2, PM5, PP3 |
| 7 | TNNT2 | thin | c.C862T | p.R288C | P | PS4, PP1, PS3, PM2, PM5, PP2 |
| 8 | MYBPC3 | THICK | c.G532A | p.V178M | LP | PM1, PM2, PP3 (strong) |
| 9 | MYL3 | thin | c.G447A | p.M149I | P | PS4, PS1, PM5, PM1, PP2, PM2, PP3 |
| 10 | TNNT2 | thin | c.C418T | p.R140C | P | PS4, PP1, PS3, PM1, PP2, PM2, PP3 |
| 11 | MYH7 | THICK | c.G3346A | p.E1116K | LP | PM2, PP3 (strong), PP2, PP5 |
| 12 | MYBPC3 | THICK | c.1351+2T>C | P | PS4, PVS1, PM2 | |
| 13 | MYBPC3 | THICK | c.3192dupC | p.K1065Qfs * 12 | P | PS4, PP1, PVS1, PM2 |
| MYBPC3 | THICK | c.C1112G | p.P371R | LP | PM2, PM1, PP3 (strong) | |
| 14 | TNNT2 | thin | c.A803T | p.K268I | LP | PS4, PM2, PP3, PP2 |
| 15 | MYH7 | THICK | c.G2770A | p.E924K | P | PP1, PS3, PS2, PM1, PP2, PM2, PM5, PP3 |
| 16 | TPM1 | thin | c.G172C | p.D58H | LP | PP3, PM2, PP2 |
| 17 | MYBPC3 | THICK | c.C1504T | p.R502W | P | PM3, PP1, PM2, PM5, PM1, PP3 |
| 18 | MYBPC3 | THICK | c.C1789T | p.R597W | P | PS4, PM2, PM5, PP3 |
| 19 | MYBPC3 | THICK | c.3331-1G>A | P | PS4, PVS1, PM2 | |
| 20 | MYBPC3 | THICK | c.G2198T | p.R733L | LP | PM1, PM2, PM5 (strong), BP4 |
| 21 | MYH7 | THICK | c.G428A | p.R143Q | P | PS4, PP1, PM1, PP2, PM2, PM5, PP3 |
| 22 | MYBPC3 | THICK | c.C1960T | p.R654C | LP | PM1, PM2, PM5 (strong), BP5 |
| 23 | MYH7 | THICK | c.A1615C | p.M539L | P | PS4, PM1, PP2, PM2, PM5, PP3 |
| 24 | MYBPC3 | THICK | c.1458-1G>A | P | PS4, PVS1, PM2 | |
| 25 | MYBPC3 | THICK | c.506-2A>C | P | PS4, PP1, PS3, PVS1, PM2 | |
| 26 | TNNT2 | thin | c.C862T | p.R288C | P | PS4, PP1, PS3, PM2, PM5, PP2 |
| 27 | MYBPC3 | THICK | c.G772A | p.E258K | P | PP3, PP5, PM2, PP2 |
| 28 | MYBPC3 | THICK | c.2943_2947del | p.Q981Hfs * 67 | P | PS4, PVS1, PM2 |
| 29 | MYBPC3 | THICK | c.1227-13G>A | LP | PM2, PS4, PS3, PP1, PP5 | |
| 30 | MYBPC3 | THICK | c.2864_2865delCT | p.P955Rfs * 95 | P | PS4, PVS1, PM2 |
| 31 | MYH7 | THICK | c.G4402C | p.E1468Q | LP | PM2, PM5, PP3, PP2 |
| 32 | MYBPC3 | THICK | c.G2459A | p.R820Q | P | PS4, PP1, PS3, PM2, PM5, PM1, PP3 |
| 33 | MYBPC3 | THICK | c.2309-2A>G | P | PS4, PP1, PVS1, PM2 | |
| 34 | MYBPC3 | THICK | c.2157_2158delTG | p.C719X | P | PS4, PVS1, PM2 |
| 35 | MYBPC3 | THICK | c.2157_2158delTG | p.C719X | P | PS4, PVS1, PM2 |
| 36 | MYH7 | THICK | c.G2680A | p.E894K | P | PS4, PM1, PP2, PM2, PM5, PP3 |
| 37 | MYBPC3 | THICK | c.913_914del | p.F305Pfs * 27 | P | PVS1, PP5, PM2, PS4, PP1 |
| 38 | MYBPC3 | THICK | c.G772A | p.E258K | LP | PS4, PP1, PS3, PM2, PP3 |
| 39 | MYBPC3 | THICK | c.G1828C | p.D610H | LP | PM1, PM2, PP3, PM5 |
| 40 | MYBPC3 | THICK | c.C1789T | p.R597W | P | PS4, PM2, PM5, PP3 |
| 41 | MYH7 | THICK | c.T1228C | p.Y410H | LP | PM1, PP2, PM2, PP3 |
| 42 | MYH7 | THICK | c.G428A | p.R143Q | P | PS4, PP1, PM1, PP2, PM2, PM5, PP3 |
| 43 | MYBPC3 | THICK | c.G1624C | p.E542Q | P | PM3, PP1, PS3, PM2, PP3 |
| 44 | MYH7 | THICK | c.C3133T | p.R1045C | P | PS4, PP3, PM2, PM5, PP2 |
| TNNT2 | thin | c.A659T | p.K220M | LP | PM1, PP2, PM2, PP3 | |
| 45 | MYBPC3 | THICK | c.913_914del | p.F305Pfs * 27 | P | PVS1, PP5, PM2, PS4, PP1 |
| MYH7 | THICK | c.G2012A | p.R671H | P | PS4, PM1, PM2, PP2, PM5, PP3 | |
| 46 | MYBPC3 | THICK | c.2258dupT | p.K754Efs * 79 | P | PS4, PVS1, PM2, PM5 |
| 47 | MYBPC3 | THICK | c.C3811T | p.R1271X | P | PM3, PS3, PVS1, PM2 |
| 48 | MYBPC3 | THICK | c.2258dupT | p.K754Efs * 79 | P | PS4, PVS1, PM2, PM5 |
| 49 | MYBPC3 | THICK | c.2157_2158delTG | p.C719X | P | PS4, PVS1, PM2 |
| 50 | MYBPC3 | THICK | c.G1505A | p.R502Q | P | PS4, PM2, PM5, PM1, PP3 |
| 51 | MYBPC3 | THICK | c.339delC | p.T114Lfs * 45 | LP | PVS1, PM2 |
| 52 | TNNT2 | thin | c.C418T | p.R140C | P | PS4, PP1, PS3, PM1, PP2, PM2, PP3 |
| 53 | TNNT2 | thin | c.C341T | p.A114V | LP | PM1, PP2, PM2, PM5, PP3 |
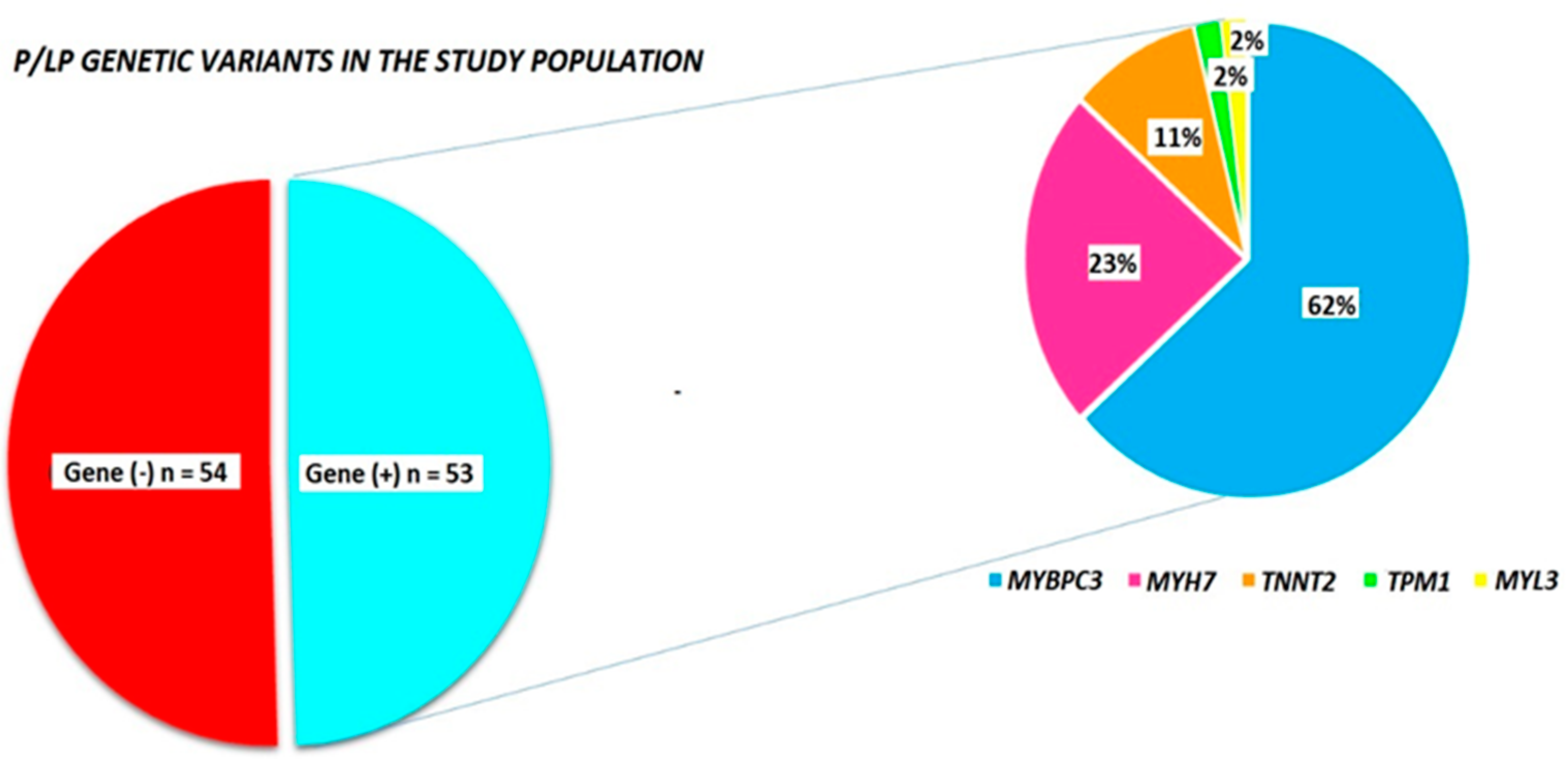
| All Pts (n = 107) | Gene (+) Pts (n = 53) | Gene (−) Pts (n = 54) | p Value | |
|---|---|---|---|---|
| Age (years) | 54 ± 16 | 50 ± 16 | 59 ± 16 | 0.01 |
| Female (%) | 42 (40%) | 26 (42%) | 20 (37%) | 0.29 |
| BMI (kg/m2) | 26.4 ± 4.4 | 26.2 ± 5.3 | 26.5 ± 3.3 | 0.51 |
| Diabetes Mellitus | 7 (7%) | 3 (6%) | 4 (7%) | 0.73 |
| Hypertension | 52 (49%) | 16 (30%) | 32 (59%) | 0.05 |
| Active smoke | 18 (17%) | 11 (21%) | 7 (13%) | 0.56 |
| Dyslipidemia | 60 (57%) | 28 (54%) | 32 (59%) | 0.57 |
| ICD, primary prevention | 10 (16%) | 5 (9%) | 6 (11%) | 0.31 |
| THERAPY (% of pts) | ||||
| β-Blockers | 81 (74%) | 244 (83%) | 37 (69%) | 0.08 |
| Dysopiramide | 5 (5%) | 4 (8%) | 1 (4%) | 0.37 |
| ACEi/ARB | 34 (36%) | 16 (37%) | 18 (35%) | 0.72 |
| Ca++Blockers | 11 (11%) | 5 (10%) | 7 (13%) | 0.59 |
| MRA | 7 (8%) | 5 (10%) | 3 (6%) | 0.63 |
| Amiodarone | 4 (4%) | 2 (4%) | 2 (4%) | 0.90 |
| Diuretics | 5 (5%) | 2 (6%) | 2 (4%) | 0.25 |
| All Pts (n = 107) | Gene (+) Pts (n = 53) | Gene (−) Pts (n = 54) | p Value | |
|---|---|---|---|---|
| Maximum wall thickness (mm) | 19 ± 5 | 19 ± 5 | 18 ± 4 | 0.21 |
| Indexed LA Volume (mL/m2) | 46 ± 16 | 49 ± 19 | 43 ± 12 | 0.04 |
| LVEF (%) | 64 ± 8 | 63 ± 9 | 64 ± 8 | 0.73 |
| E/e’ | 11.1 ± 5.0 | 11.0 ± 4.5 | 11.3 ± 5.2 | 0.55 |
| GLS (%) | −15.7 ± 4.1 | −16.6 ± 4.4 | −15.0 ± 3.7 | 0.09 |
| Mitral regurgitation, mild to moderate | 81 (76%) | 43 (83%) | 38 (70%) | 0.18 |
| LVTO (rest or Valsalva manoeuvre) | 25 (23%) | 8 (15%) | 17 (31%) | 0.08 |
| All Pts (n = 107) | Gene (+) Pts (n = 53) | Gene (−) Pts (n = 54) | p Value | |
|---|---|---|---|---|
| Peak RER | 1.11 ± 0.15 | 1.12 ± 0.18 | 1.10 ± 0.10 | 0.39 |
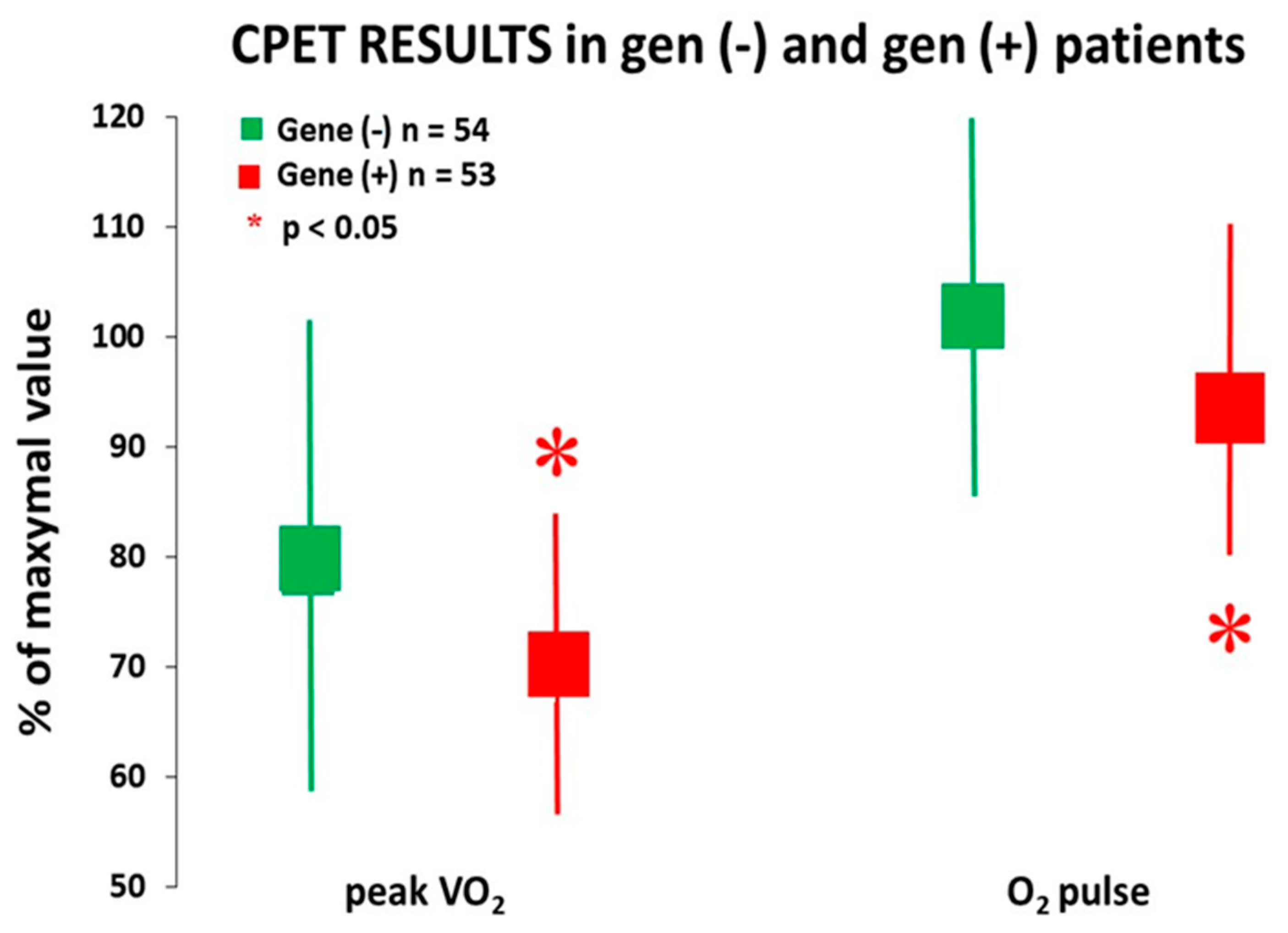
| Peak VO2 (mL/kg/min) | 20.5 ± 7.5 | 20.1 ± 7.5 | 20.3 ± 7.0 | 0.94 |
| Peak VO2 (% predicted) | 77.0 ± 19.5 | 73.0 ± 16.8 | 80.0 ± 21.5 | 0.05 |
| AT VO2 (% VO2 max) predicted) | 52 ± 19 | 50 ± 21 | 52 ± 18 | 0.59 |
| O2 pulse peak (mL/beat) | 13.1 ± 3.7 | 13.0 ± 3.2 | 13.3 ± 4.1 | 0.25 |
| O2 pulse peak (% predicted) | 99.0 ± 21.4 | 96.5 ± 17.9 | 103.0 ± 23.7 | 0.03 |
| VO2/Work [(mL/kg/min)]/Watts | 9.7 ± 2.3 | 9.4 ± 1.8 | 10.0 ± 2.5 | 0.16 |
| VE/VCO2 slope | 31.2 ± 6.3 | 32.1 ± 7.0 | 30.7 ± 5.5 | 0.37 |
| P(ET)CO2 (mmHg) | 32.0 ± 5.2 | 31.2 ± 6.6 | 33.5 ± 4.6 | 0.06 |
| All Gene (+) Pts (n = 52) | Gene (+THICK) Pts (n = 45) | Gene (+Thin) Pts (n = 7) | p Value | |
|---|---|---|---|---|
| Age (years) | 50.1 ± 14.6 | 49.4 ± 15.5 | 51.2 ± 11.7 | 0.71 |
| Female (%) | 22 (42%) | 20 (44%) | 2 (28%) | 0.21 |
| BMI (kg/m2) | 26.3 ± 5.5 | 26.0 ± 5.2 | 27.1 ± 2.9 | 0.81 |
| Diabetes Mellitus | 3 (6%) | 2 (4%) | 1 (14%) | 0.38 |
| Hypertension | 20 (38%) | 17 (39%) | 3 (43%) | 0.95 |
| Active smoke | 11 (21%) | 8 (18%) | 3 (43%) | 0.05 |
| Dyslipidemia | 28 (54%) | 23 (52%) | 5 (63%) | 0.59 |
| ICD, primary prevention | 7 (13%) | 6 (14%) | 1 (13%) | 0.93 |
| THERAPY (% of pts) | ||||
| β-Blockers | 42 (81%) | 35 (78%) | 7 (100%) | 0.22 |
| Dysopiramide | 4 (8%) | 4 (9%) | none | - |
| ACEi/ARB | 20 (38%) | 15 (33%) | 5 (71%) | 0.11 |
| Ca++Blockers | 5 (10%) | 3 (7%) | 2 (25%) | 0.11 |
| MRA | 5 (10%) | 3 (7%) | 2 (9%) | 0.11 |
| Amiodarone | 2 (4%) | 2 (5%) | none | - |
| Diuretics | 3 (6%) | 2 (5%) | 1 (13%) | 0.37 |
| Gene (+) Pts (n = 52) | Gene (+Thick) Pts (n = 45) | Gene (+Thin) Pts (n = 7) | p Value | |
|---|---|---|---|---|
| Maximum wall thickness (mm) | 19.7 ± 5.1 | 20.3 ± 5.1 | 16.5 ± 3.4 | 0.04 |
| Indexed LA Volume (mL/m2) | 48.1 ± 15.4 | 47.5 ± 15.7 | 51.5 ± 14.3 | 0.50 |
| LVEF (%) | 63.3 ± 8.6 | 63.9 ± 7.8 | 60.0 ± 12.2 | 0.24 |
| E/e’ | 10.9 ± 4.7 | 10.8 ± 4.8 | 11.38 ± 4.0 | 0.74 |
| GLS (%) | −15.92 ± 4.5 | −16.32 ± 4.6 | −12.8 ± 3.3 | 0.15 |
| Mitral regurgitation, mild to mild-moderate | 41 (79%) | 35 (78%) | 6 (86%) | 0.31 |
| Mild LVTO at rest or after Valsalva manoeuvre | 4 (8%) | 4 (9%) | none | 0.81 |
| All Gene (+) Pts (n = 52) | Gene (+THICK) Pts (n = 45) | Gene (+Thin) Pts (n = 7) | p Value | |
| Peak RER | 1.11 ± 0.12 | 1.12 ± 0.11 | 1.10 ± 0.16 | 0.21 |
| Peak VO2 (mL/kg/min) | 20.1 ± 8 | 20.9 ± 7.6 | 16.3 ± 2.7 | 0.12 |
| Peak VO2 (% predicted) | 70.5 ± 18.3 | 74.2 ± 15.6 | 58.6 ± 10.8 | 0.01 |
| AT VO2 (% predicted) | 49.7 ± 21.0 | 49.8 ± 15.3 | 40.9 ± 10.9 | 0.15 |
| O2 pulse peak (mL/beat) | 12.8 ± 3.56 | 13.4 ± 9.9 | 11.2 ± 2.3 | 0.60 |
| O2 pulse peak (% predicted) | 94.5 ± 11.5 | 97.8 ± 17.0 | 82.2± 9.9 | 0.02 |
| VO2/Work [(mL/kg/min)]/Watts | 9.3 ± 1.8 | 9.5 ± 1.6 | 8.3 ± 1.3 | 0.05 |
| VE/VCO2 slope | 31.3 ± 6.9 | 31.7 ± 6.5 | 30.6 ± 8.2 | 0.96 |
| P(ET)CO2 (mmHg) | 31.6 ± 7.7 | 32.0 ± 7.9 | 30.9 ± 9.2 | 0.61 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, M.F.; Malfatto, G.; Baratto, C.; Giglio, A.; Rella, V.; Cerea, P.; Mariani, D.; Salerno, S.; Ravaro, S.; Castelletti, S.; et al. The Influence of Genotype on the Cardiopulmonary Test Response in Patients Affected by Hypertrophic Cardiomyopathy. Cardiogenetics 2025, 15, 12. https://doi.org/10.3390/cardiogenetics15020012
Gagliardi MF, Malfatto G, Baratto C, Giglio A, Rella V, Cerea P, Mariani D, Salerno S, Ravaro S, Castelletti S, et al. The Influence of Genotype on the Cardiopulmonary Test Response in Patients Affected by Hypertrophic Cardiomyopathy. Cardiogenetics. 2025; 15(2):12. https://doi.org/10.3390/cardiogenetics15020012
Chicago/Turabian StyleGagliardi, Maria Felicia, Gabriella Malfatto, Claudia Baratto, Alessia Giglio, Valeria Rella, Paolo Cerea, Davide Mariani, Sabrina Salerno, Silvia Ravaro, Silvia Castelletti, and et al. 2025. "The Influence of Genotype on the Cardiopulmonary Test Response in Patients Affected by Hypertrophic Cardiomyopathy" Cardiogenetics 15, no. 2: 12. https://doi.org/10.3390/cardiogenetics15020012
APA StyleGagliardi, M. F., Malfatto, G., Baratto, C., Giglio, A., Rella, V., Cerea, P., Mariani, D., Salerno, S., Ravaro, S., Castelletti, S., Fratianni, G., Alberio, C., Pedrazzini, M., Khujadze, M., Badano, L. P., Muraru, D., Parati, G., Cecchi, F., Caravita, S., & Crotti, L. (2025). The Influence of Genotype on the Cardiopulmonary Test Response in Patients Affected by Hypertrophic Cardiomyopathy. Cardiogenetics, 15(2), 12. https://doi.org/10.3390/cardiogenetics15020012