



Cardiogenetics 2026, 16(2), 10; https://doi.org/10.3390/cardiogenetics16020010 - 11 May 2026
Abstract
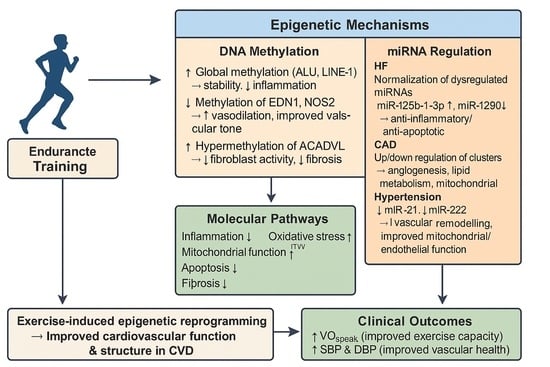
Background: Genetic testing in hypertrophic cardiomyopathy (HCM) yields variable positivity rates. Identifying clinical predictors of positive genetic tests could improve pre-test counseling and refine expectations about diagnostic yield. Methods: We analyzed consecutive genotyped HCM probands from a contemporary multicenter cohort across four Italian
[...] Read more.
Background: Genetic testing in hypertrophic cardiomyopathy (HCM) yields variable positivity rates. Identifying clinical predictors of positive genetic tests could improve pre-test counseling and refine expectations about diagnostic yield. Methods: We analyzed consecutive genotyped HCM probands from a contemporary multicenter cohort across four Italian tertiary centers. Genotype positivity was defined as the presence of ≥1 pathogenic or likely pathogenic variant (ACMG classes 4–5). Multivariable logistic regression identified predictors of genotype positivity. Sensitivity analyses assessed the incremental value of left atrial volume index (LAVI) ≥ 34 mL/m2 and the mode of first clinical presentation. Results: Among 274 genotyped probands (median age at diagnosis 54 years; 62% male), 86 (31%) were genotype-positive (38% MYBPC3, 29% MYH7). Age at diagnosis <40 years (OR 2.38, 95%CI 1.26–4.51, p = 0.008), family history of sudden cardiac death/major ventricular arrhythmias (OR 2.34, 95%CI 1.16–4.84, p = 0.019) and family history of non-ischemic cardiomyopathy (OR 1.92, 95%CI 1.04–3.54, p = 0.038), were independently associated with genotype positivity whereas arterial hypertension was inversely associated (OR 0.42, 95%CI 0.23–0.77). Maximal left ventricular wall thickness > 20 mm and gender were not predictive of genotype positivity. Inclusion of LAVI modestly improved the model performance (AUC 0.769, p = 0.016, ΔAUC +0.024; DeLong p = 0.016) but without leading to meaningful patient reclassification. Conclusions: Genotype positivity in HCM links to earlier onset and family history; traditional severity markers and initial presentation may not independently suggest genetic causality. These findings may help shape a personalized approach to genetic counseling in HCM.
Full article
(This article belongs to the Special Issue Contemporary and Future Approaches to Inherited Cardiomyopathies)
►
Show Figures

Graphical abstract





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}