



Dermatopathology 2026, 13(3), 31; https://doi.org/10.3390/dermatopathology13030031 - 3 Jul 2026
Abstract
Background: Melanoma incidence is rising rapidly worldwide and stands out due to its high lethality. Despite advances in clinical treatment and in understanding melanoma-sensitive genes and molecular pathogenesis, a specific area of ongoing research is the connection between stress-related β-adrenergic receptors (ARs), hypoxia,
[...] Read more.
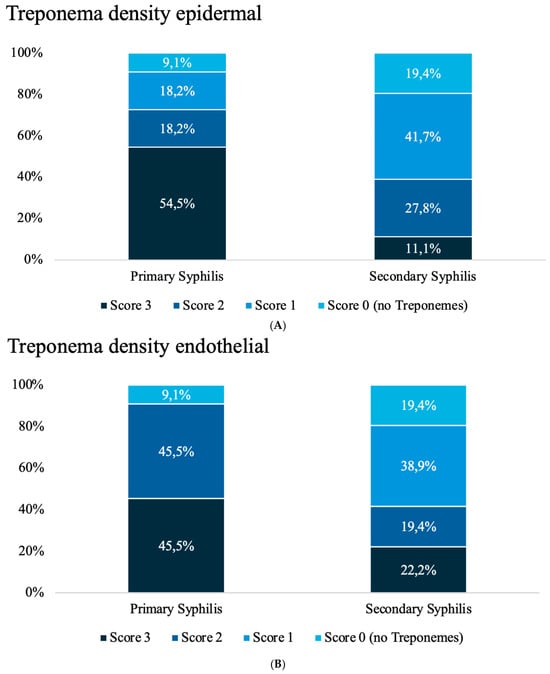
Background: Melanoma incidence is rising rapidly worldwide and stands out due to its high lethality. Despite advances in clinical treatment and in understanding melanoma-sensitive genes and molecular pathogenesis, a specific area of ongoing research is the connection between stress-related β-adrenergic receptors (ARs), hypoxia, and neovascularization in melanoma tumor progression. This exploratory study aimed to investigate the expression of β3-AR, HIF-1α, and CD31 in several cellular subsets of atypical melanocytic lesions and their interplay in promoting melanoma malignancy. Methods: Twenty-seven patients with melanocytic lesions at different stages that were surgically removed were retrospectively selected; clinical-pathological and dermoscopic data were collected. Results: Immunohistochemical and digital evaluation revealed a significant upregulation of β3-AR in malignant melanoma melanocytes and in macrophages from invasive >pT1a melanomas compared to dysplastic nevi. Increased HIF-1α expression in malignant melanocytes and CD31 expression levels in >pT1a melanomas were observed. Ulcerated lesions exhibited a higher percentage of β3-AR, HIF-1α, and CD31 expression. Pearson correlation analysis revealed positive associations among these markers in human malignant melanoma, suggesting a potential relationship between adrenergic signaling, hypoxia, and tumor vascularization. Conclusions: These exploratory findings suggest that β3-AR, HIF-1α, and CD31 may represent interconnected components of the melanoma microenvironment. Unraveling these interactions in larger, independent cohorts and functional studies may provide additional insights into melanoma biology and help define their potential translational relevance.
Full article
(This article belongs to the Section Experimental Dermatopathology)






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}