Abstract
Myo-inositol (MI) is an essential dietary polyol involved in host metabolism. However, the identity and diversity of MI-utilizing gut bacteria in poultry remain largely unknown. This study aimed to isolate anaerobically growing bacteria enriched under MI-based cultivation conditions from two commercial laying hen breeds. Digesta samples were cultured on minimal growth media containing MI as the sole (Trial 1) or principal (Trial 2) carbon source. Isolates were purified, screened by amplified ribosomal DNA restriction analysis (ARDRA), and identified using 16S rRNA gene sequencing. Among 42 sequenced isolates, ten representative strains were classified within the phyla Pseudomonadota (n = 5), Bacillota (n = 4), and Bacteroidota (n = 1). Members of the Escherichia-Shigella clade were most frequently recovered, followed by Clostridium, Enterococcus, Pediococcus, and Bacteroides. Selected Escherichia-Shigella isolates were screened negative for ipah and ial virulence genes, except for three isolates that tested positive only for the ipah gene. These findings provide the first culture-based framework for investigating MI-responsive bacteria in the chicken gut.
1. Introduction
Myo-inositol (MI), a cyclo-hexitol sugar alcohol, is the most abundant form of inositol and plays a significant role in avian physiology, particularly in metabolic regulation and cellular functions [1,2]. MI is ubiquitously present in plant- and animal-derived feed and functions as a key osmolyte, a precursor of secondary messengers, and a structural component of membrane phospholipids in eukaryotic cells [3,4]. In poultry, MI has been associated with modulation of insulin sensitivity, regulation of circadian rhythms, calcium signaling, brain function, lipid metabolism, behavior, and production performance [1,5,6,7]. Despite these physiological roles, the catabolism of this polyol within the poultry gastrointestinal tract remains incompletely understood. Specifically, little is known about the identity or diversity of gut microbes capable of utilizing MI as a carbon source.
Most poultry nutrition studies focus on phytate degradation, where MI appears as a terminal hydrolysis product in the gastrointestinal tract. However, the fate of MI beyond its release from phytate, particularly its microbial utilization, has not been fully explored. Breed-specific differences have been reported, with higher MI concentration in the ileum of Lohman Brown-classic (LB) hens compared to Lohmann LSL-classic (LSL) hens, suggesting a host genetic influence on MI metabolism [8]. Investigations into electrogenic MI transport in the small intestine revealed no detectable uptake or translocation across the epithelium despite the presence of known transporters (SMIT-1 and SMIT-2), implying that microbial pathways may contribute substantially to MI metabolism [9,10].
While MI biosynthesis has been studied extensively in microorganisms such as Candida spp., Saccharomyces cerevisiae, and Escherichia coli (E. coli) via genetic engineering or adaptive evolution approaches [11,12], MI catabolism has been documented in a more limited number of bacterial species. These include Aerobacter aerogenes, Lactobacillus casei, Bacillus subtilis, Salmonella enterica, and Enterobacter spp. YB-46, Anaerostipes rhamnosivorans, Mitsuokella jalaludinii, Megamonas rupellensis, Blautia schinkii, and Dysosmobacter welbionis, in which MI has been shown to support growth or be metabolically converted under defined conditions [4,10,13,14,15,16,17,18,19,20,21]. Among these, only Anaerostipes, Mitsuokella, Megamonas, and Dysosmobacter have been experimentally shown to metabolize MI under anaerobic conditions, highlighting the limited understanding of anaerobic MI degradation in gut bacteria.
Recent evidence from anaerobic gut microbes highlights that MI can serve as a substrate for anaerobic metabolism and microbial cross-feeding in complex communities. For example, Dysosmobacter welbionis and Blautia schinkii in the human gut can anaerobically utilize MI [19,21], while culture-based work in pigs has demonstrated that MI released during phytate degradation is shared among phytase-producing and MI-utilizing strains [22]. Together, these findings emphasize the potential role of MI as a shared microbial substrate, but comparable culture-based evidence in the poultry gut remains scarce, justifying the focus of the present study on isolating and characterizing potentially MI-utilizing anaerobes.
A genome-wide analysis further indicates that inositol catabolic gene clusters (IolCatGCs) are unevenly distributed among gut-associated bacterial taxa, with representatives across diverse phyla such as Bacillota, Pseudomonadota, and Bacteroidota [23]. This suggests that MI utilization is likely a strain-specific trait and may confer ecological advantages or niche specialization in the gut microbiome.
Given the functional relevance of MI and the limited knowledge of its microbial degradation in the poultry gut, this study aimed to isolate and characterize anaerobically growing bacteria from the gastrointestinal tracts of LB and LSL laying hens using MI-enriched selective media. By combining culture-based enrichment with 16S rRNA gene sequencing, we sought to identify bacterial taxa with a potential MI-metabolizing capacity. This approach not only expands the catalog of bacteria with anaerobic MI-degradation potential but also provides viable cultures for subsequent genomic, transcriptomic, and functional analyses. Understanding the microbial contribution to MI turnover could ultimately inform strategies to enhance nutrient utilization and gut health in commercial poultry production.
2. Materials and Methods
2.1. Experimental Design and Animal Sampling
This study was part of the interdisciplinary Research Unit P-Fowl: Inositol phosphates and myo-inositol in the domestic fowl: Exploring the interface of genetics, physiology, microbiome, and nutrition (project homepage: https://p-fowl.uni-hohenheim.de/en, accessed on 12 February 2026). In this study, two independent trials were conducted using LSL and LB laying hens housed at the Agricultural Experimental Station of the University of Hohenheim at Unterer Lindenhof, Eningen, Germany.
Feed and tap water were given ad libitum. In Trial 1, hens received the MI3 diet, a corn and soybean meal-based diet supplemented with 3 g/kg MI (full composition published in Sommerfeld et al. 2025 [2]). Trial 2 hens received a corn and soybean meal-based diet formulated to meet all nutrient requirements except phosphorus (full composition is listed in Supplementary Table S1).
On sampling day, hens were anesthetized using a controlled gas mixture (35% CO2, 35% N2, and 30% O2) and euthanized by immediate decapitation. In Trial 1, two hens per breed were sacrificed, and in Trial 2, four hens per breed were sacrificed. To capture the diversity of microbial populations potentially involved in MI metabolism, samples were specifically obtained from the crop, duodenum, ileum and caeca sections in Trial 1 and from the ileum in Trial 2 (Figure 1). Each section was aseptically excised and transferred into sterile, N2-flushed flasks containing transport media (sodium thioglycolate broth) supplemented with resazurin as a redox indicator, ensuring reduced and anaerobic conditions (full composition in Supplementary Table S2). Samples were maintained at 35–40 °C during the 3–4 h transport and processed immediately upon arrival.
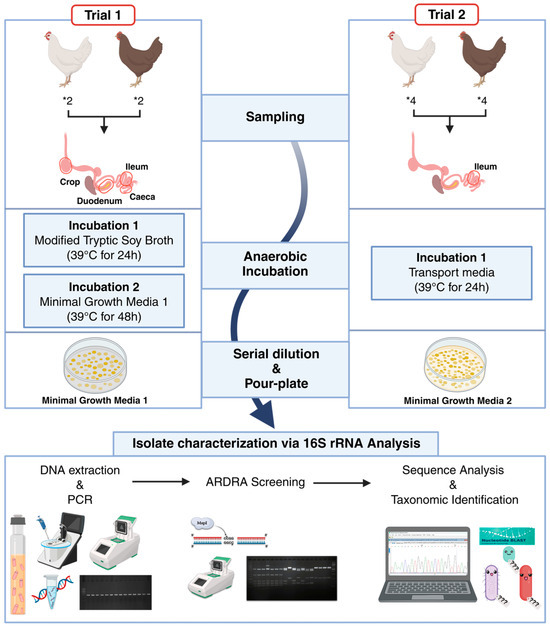
Figure 1.
Schematic overview of the experimental design used to isolate and identify potential myo-inositol-utilizing bacteria from the gastrointestinal tracts of laying hens. The workflow illustrates sample collection from different intestinal compartments in Trial 1 and the ileum in Trial 2, anaerobic enrichment and cultivation using MI-supplemented media, isolation, DNA extraction and Polymerase Chain Reaction (PCR) amplification, dereplication by Amplified Ribosomal DNA Restriction Analysis (ARDRA), and taxonomic identification by partial 16S rRNA gene sequencing. * indicates a multiplication sign in a quantity. Created in BioRender. Seifert, J. (2026) https://BioRender.com/x5fpy1l, accessed on 9 February 2026.
2.2. Enrichment and Cultivation
Two experimental trials were conducted using a custom-designed minimal growth medium to selectively enrich MI-utilizing bacteria. The primary selective pressure in both trials was the inclusion of MI as a defined carbohydrate source. In Trial 1, MI (1 g/L) served as the sole carbon source (minimal growth media 1). In Trial 2, although tryptone (15 g/L) and soya peptone (3 g/L) provided carbon in the form of amino acids and peptides, MI (3.6 g/L) represented the major readily usable carbon source, providing the principal substrate for microbial growth (minimal growth media 2).
Upon arrival in the laboratory, the samples were further handled in an anaerobic station (Whitley A35 workstation, Don Whitley Scientific, Bingley, UK) that contained a mixture of 80% N2 (quality level 5.0), 15% CO2 (quality level 3.0) and 5% H2 (quality level 5.0) and using sterile scissors and spatula, the sections were cut open and the digesta material was transferred to the respective media [24]. For each trial, digesta samples from laying hens of the same breed and gut compartment were pooled prior to enrichment. Specifically, in Trial 1, digesta from two LB and two LSL hens were collected per gut compartment (crop, duodenum, ileum, and caeca) and pooled by breed and compartment before inoculation into enrichment media. In Trial 2, digesta from birds of the same breed were similarly pooled prior to enrichment. A single anaerobic enrichment culture was established for each pooled sample, and no technical replicates were performed.
In Trial 1, digesta was directly transferred to a modified tryptic soy broth (TSB) in which glucose was replaced with MI as the sole added carbohydrate source to provide a nutrient-rich but MI-selective environment for initial enrichment. Cultures were incubated at 39 °C for 24 h, and then transferred to minimal growth medium 1 and incubated for a further 48 h at 39 °C. In Trial 2, the digesta was returned to the transport medium for 24 h. Although the internal body temperature of chickens ranges between 40 and 42 °C, many culture-based studies on poultry gut microbiota employ incubation temperatures around 37 °C to efficiently recover mesophilic gut microbiota without imposing thermal stress [25]. In this study, incubation was performed at 39 °C, balancing proximity to the host’s internal temperature with optimal growth conditions for mesophilic gut bacteria, consistent with temperatures used in a previous poultry isolation study [24]. The detailed composition of all media used during both trials is listed in Supplementary Table S2.
2.3. Isolation, Purification and Preservation
Anaerobically growing bacteria were isolated from the enriched cultures using standard serial dilution in sterile physiological solution (0.85% NaCl). Samples were serially diluted over a range of 10−1 to 10−6, and 1 mL aliquots of selected dilutions were pour-plated onto the corresponding minimal agar media. For isolation, plates corresponding to dilutions yielding well-separated colonies were selected for downstream purification. In Trial 1, cultures from minimal growth medium 1 were plated on minimal growth medium 1 agar, whereas in Trial 2, cultures from 24 h of incubation in transport medium were plated on minimal growth medium 2 agar. Plates were incubated anaerobically at 39 °C for 1–4 days and monitored until colonies appeared.
Individual, well-isolated colonies displaying distinct morphologies were picked and subjected to multiple rounds of purification using the streak plate method on the respective media to obtain axenic cultures. Purity was assessed based on consistent recovery of uniform colony morphology across repeated subculturing steps.
Purified isolates were preserved as glycerol stocks by mixing equal volumes of actively growing culture and sterile 50% (v/v) glycerol solution to obtain a final glycerol concentration of 25% (v/v). The suspensions were homogenized by vortexing and stored at ultra-low temperatures of −80 °C to maintain viability and genetic integrity for downstream molecular characterization, following the method described by Naithani et al. 2024 [24].
2.4. Molecular Characterization
Different isolated pure bacterial colonies streaked and incubated for 24 h in respective agar plates were suspended in sterile phosphate-buffered saline (PBS) solution and washed twice. Genomic DNA was extracted from the pure, isolated cultures using the QIAamp PowerFecal Pro DNA kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions, quantified using a Nanodrop spectrophotometer, and stored at −20 °C. The extracted DNA served as the template for Polymerase Chain Reaction (PCR) amplification of the 16S rRNA gene. DNA concentration was set between 15 and 100 ng/µL, and universal primers 27f (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492r (5′-TACGGYTACCTTGTTACGACTT-3′) were used to amplify the 16S rRNA region [26]. Thermocycling was performed on a Bio-Rad T100 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) with the following program: initial denaturation at 98 °C for 3 min, 30 cycles of denaturation at 98 °C for 10 s, annealing at 55 °C for 15 s, and extension at 72 °C for 3 min, followed by a final extension at 72 °C for 10 min and a hold at 4 °C. The quality of the PCR product was checked by 1.5% agarose gel electrophoresis.
2.5. Amplified Ribosomal DNA Restriction Analysis (ARDRA)
ARDRA was performed on the 16S rDNA gene PCR products to assess genetic diversity and reduce redundancy among the isolates. PCR products were digested with the restriction enzymes MspI (NEB Inc., Ipswich, MA, USA) under the following conditions: 37 °C for 1 h with the thermocycler (T100, Bio-Rad Laboratories, USA) lid set to 105 °C, followed by an indefinite hold at 4 °C. Digested fragments were visualized on 2.5% agarose gels to generate distinct restriction fragment length polymorphism (RFLP) patterns [27].
2.6. 16S rRNA Gene Sequencing and Taxonomic Identification
Based on RFLP patterns, PCR products of selected isolates were purified using the MinElute® PCR purification kit (Qiagen) as per the manufacturer’s instructions. The purified PCR products were sent for single-direction Sanger sequencing of the 16S rRNA gene (Microsynth Seqlab GmbH, Göttingen, Germany).
Raw 16S rRNA gene sequences were initially obtained using either the forward or reverse primer (see Supplementary Table S4). Sequences were inspected for quality and trimmed to remove low-quality base calls using MEGA version 12 [28]. For preliminary taxonomic identification, high-quality sequences (800–1200 bp) were compared against the NCBI “16S ribosomal RNA sequences (Bacteria and Archaea)” database using online NCBI BLASTn [29]. Species-level identification was assigned to sequences with more than 99% percentage identity and more than 98% query coverage to a reference sequence. In case of getting similar hits, preference was given to type strains, RefSeq curated sequences, and completeness of the reference sequence.
Isolates producing sequences with identical top BLASTn hits (i.e., matching NCBI accession numbers) were grouped to identify unique strains. One representative isolate from each group was subsequently sequenced using the complementary primer to obtain near full-length 16S rRNA gene sequences (1000–1500 bp). Sequences were subsequently trimmed, aligned using MEGA version 12, and compared again against the NCBI “16S ribosomal RNA sequences (Bacteria and Archaea)” database (accessed on 20 October 2025) using the online NCBI BLASTn tool for final taxonomic confirmation [28,29]. The 16S rRNA gene sequences of the representative isolates were submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena, accessed on 22 February 2026).
2.7. Risk Group Assessment
The biosafety classification of the identified isolates was determined according to the Technical Rules for Biological Agents (TRBA 466) issued by the Federal Ministry of Labour and Social Affairs [30]. This guideline categorizes microorganisms into Risk Groups (RG) 1–4, based on their pathogenic potential, transmissibility, and the availability of effective prophylaxis or therapeutic interventions.
Taxonomic assignments obtained from 16S rRNA gene sequencing were cross-referenced with the TRBA 466 list to determine the RG classification of each isolate. Additional annotations provided in the TRBA framework (e.g., toxin-production or vertebrate (animal/human) pathogenic potential) were also recorded where applicable. This assessment was performed solely to ensure appropriate laboratory handling and biosafety compliance and does not imply pathogenic or ecological functions of the isolates.
2.8. Screening for Virulence Genes of Selected Isolates
As part of a preliminary biosafety assessment, a subset of 13 selected DNA samples from isolates belonging to the Escherichia-Shigella clade were subjected to PCR screening to assess potential pathogenicity and to discriminate between Shigella and enteroinvasive E. coli (EIEC).
The virulence genes targeted were ipaH (invasion plasmid antigen H) and ial (invasion-associated locus), both of which are commonly associated with the pathogenicity of Shigella and EIEC. Primer sequences and expected fragment lengths were: ipaH-F/R (GTT CCT TGA CCG CCT TTC CGA TAC CGTC/GCC GGT CAG CCA CCC TCT GAG AGT AC, 619 bp), detecting both Shigella and EIEC; ipaH-Shig1/2 (TGG AAA AAC TCA GTG CCT CT/CCA GTC CGT AAA TTC ATT CT, 423 bp), specific for Shigella; and ial-F/R (CTG GAT GGT ATG GTG AGG/GGA GGC CAA CAA TTA TTT CC, 320 bp), detecting the invasion-associated locus [31,32].
PCR amplification was performed under the following cycling conditions: initial denaturation at 95 °C for 5 min, followed by 30 cycles of denaturation at 95 °C for 15 s, annealing at 50 °C for 1 min, and extension at 72 °C for 1 min, with a final extension at 72 °C for 5 min. Further, the amplified products were analyzed by agarose gel electrophoresis alongside a 250–1500 bp DNA ladder and an appropriate positive control for interpretive rigor.
3. Results
3.1. Selection of Representative Isolates
Distinct colony morphologies were observed after incubation on minimal growth media, indicating successful enrichment of bacteria capable of growth under MI-supplemented conditions. Across both trials, 107 isolates were recovered from the different intestinal compartments (numbers per compartment and breed are provided in Supplementary Table S3). ARDRA of the 16S rRNA gene PCR products revealed 17 unique RFLP patterns in total, with 11 patterns originating from Trial 1 and 6 from Trial 2.
To capture potential strain-level variation and ensure comprehensive representation, a total of 42 representative isolates were selected for further analysis. This set included all unique ARDRA patterns as well as a subset of isolates with similar patterns but originating from different intestinal segments, breeds, or trials. These additional isolates were sequenced to verify that identical RFLP profiles corresponded to the same taxonomic identity across biological sources and to capture any potential intra-pattern variation.
3.2. Overall Taxonomic Diversity
In Trial 1, a total of 63 colonies were picked, and 21 isolates were selected for sequencing based on RFLP patterns. From these, 14 isolates originated from LB hens (two from crop, five from duodenum, four from ileum, and three from caeca) and seven from LSL hens (three from duodenum, three from ileum, and one from caeca). Out of these, 19 belonged to phylum Pseudomonadota, one to Bacillota, and one to Bacteroidota. In Trial 2, 44 colonies were picked, screened with ARDRA, and 21 selected isolates were sequenced (ten from LB hens and eleven from LSL hens). Out of these, 15 are members of Bacillota and six of Pseudomonadota.
Overall, 42 (21 from each trial) bacterial isolates were identified using partial 16S rRNA gene sequences (800–1200 bp) (Figure 2). BLASTn analysis revealed high-confidence matches, with query coverage ranging from 95 to 100%, percent identities between 98.9 and 100%, bit scores exceeding 1400, and E-values of 0, indicating statistically significant alignments. Top matches, accession numbers, and other details are summarized in Table 1 along with isolate source information.
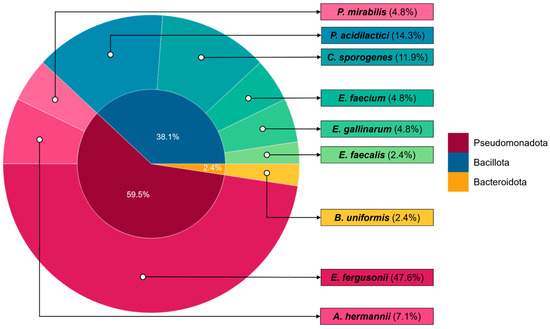
Figure 2.
Taxonomic distribution of all bacterial isolates recovered in this study based on partial 16S rRNA gene sequencing. The inner circle represents the relative distribution of isolates at the phylum-level, while the outer circle shows the corresponding genera identified within each phylum. The figure includes isolates obtained from both experimental trials and all sampled intestinal regions, providing an overview of the diversity of potential MI-utilizing gut bacteria identified in this study.

Table 1.
Overview of all bacterial isolates recovered from Lohmann Brown-classic (LB) and Lohmann LSL-classic (LSL) laying hens. The table summarizes the gut compartment of origin, experimental trial, NCBI BLASTn-based taxonomic identification derived from partial 16S rRNA gene sequences, and corresponding phylum assignment for each isolate.
3.3. Strain Diversity Analysis
Analysis of 16S rRNA gene sequences and their corresponding NCBI accession numbers showed that among the 42 isolates sequenced, only ten represented unique accession numbers, indicating a high degree of genetic relatedness at the 16S rRNA level among several isolates from different gut sections, breeds, or trials (Table 2). This suggests that while multiple colonies were recovered, a subset represented repeated isolation of the same bacterial strains. Only the unique strains were selected for further comparative analysis. The 16S rRNA gene sequences of these representative strains were deposited in ENA under accession numbers listed in Table 2.
Table 2.
Taxonomic identification of representative bacterial isolates based on partial 16S rRNA gene sequence analysis. The table includes the closest NCBI BLASTn match, percentage sequence identity, assigned taxonomic classification, phylum affiliation, and corresponding European Nucleotide Archive (ENA) accession numbers.
Notably, two groups of Escherichia fergusonii isolates were recovered from multiple gut sections with identical NCBI accession numbers. However, BLAST results of the majority of these isolates showed similar percentage identity and query coverage with both Shigella and Escherichia species. 16S rRNA-based identification for this group is ambiguous since they share high similarity [33,34]. Cross-referencing with other databases predominantly assigned these isolates to Shigella flexneri (EZBioCloud) or E. coli (GTDB), highlighting the limitations of 16S rRNA for resolving closely related members of the Enterobacteriaceae (Table S2). Accordingly, in this study, isolates belonging to the Escherichia-Shigella clade are reported at the clade level rather than at the species level.
3.4. Phylum-Level Comparison and Distribution
To assess the taxonomic breadth of the representative isolates, the ten non-redundant strains were classified at the phylum level based on 16S rRNA gene sequences. These isolates were assigned to three distinct bacterial phyla: Bacillota, Pseudomonadota, and Bacteroidota.
The majority of the isolates belonged to the phylum Bacillota (n = 5), including Clostridium, Enterococcus, and Pediococcus. This phylum was recovered by both breeds, especially from Trial 2. Pseudomonadota accounted for four of the representative isolates, including members of the Enterobacteriaceae such as Escherichia-Shigella clade and Proteus. These isolates were recovered across breeds, gut sections, and trials. Only one isolate was affiliated within the phylum Bacteroidota, identified as Bacteroides uniformis, and was obtained from the ileum of LSL in Trial 1.
3.5. Breed-Level Comparison
Analysis of the total bacterial isolates showed some variation in species composition between the two commercial laying hen breeds (LSL vs. LB). Across both trials, a total of 24 isolates were recovered from LB hens and 18 from LSL hens. Based on representative strains, both breeds showed a small set of shared species, including E. fergusonii and Pediococcus acidilactici, whereas the remaining strains were exclusive to a single breed. The unique strains isolated from LB hens were Clostridium sporogenes, Enterococcus gallinarum, and Enterococcus faecalis, all belonging to the phylum Bacillota. In contrast, the unique isolates from LSL hens were Atlantibacter hermannii, Bacteroides uniformis, Proteus mirabilis, and Enterococcus faecium, spanning the phyla Pseudomonadota, Bacteroidota, and Bacillota.
3.6. Biosafety and Regulatory Implications
The biosafety assessment, conducted according to the Technical Rules for Biological Agents [30], categorized nine out of the ten representative isolates recovered under MI selective conditions as RG 2, including members of the Escherichia-Shigella clade, Atlantibacter hermannii, Clostridium sporogenes, Enterococcus spp., Bacteroides uniformis, and Proteus mirabilis. Only Pediococcus acidilactici, a non-pathogenic lactic acid bacterium, was classified as RG 1, indicating minimal pathogenic potential and suitability for a categorization in Biosafety Level 1 (Table 3). No isolates were assigned to RG 3 or 4, confirming that this study did not introduce high-consequence pathogens into the laboratory environment. The compiled results, including the identified strain, assigned RG, and relevant remarks, are presented in Table 3. The annotations (“+”, “ht”, and “TA”) in the table further indicate biosafety-relevant traits, implicating their potential to pose moderate health hazards in laboratory environments. Given institutional biosafety restrictions, cultures were either preserved appropriately or discarded as per rules and regulations.
Table 3.
Risk group (RG) classification of representative bacterial isolates according to TRBA 466 (BAuA 2010; Classification of prokaryotes (bacteria and archaea) into RG). RG assignments and relevant remarks are provided to indicate biosafety considerations associated with each isolate.
3.7. Virulence Gene Screening
As part of a preliminary biosafety assessment, 13 selected isolates from the Escherichia-Shigella clade were screened for virulence genes. PCR amplification of ipaH, Shigella-specific ipaH-Shig1/2, and ial genes revealed that ten isolates were negative for all targets, while three isolates (BDu2, CCa1, and BCa1) tested positive only for ipaH-F/R and were negative for the other genes (Table 4; Figure S1).
Table 4.
PCR-based screening of selected isolates within the Escherichia-Shigella clade for the presence of virulence-associated genes (ipaH and ial). The table summarizes gene detection results for each isolate to assess potential pathogenic traits.
4. Discussion
MI is a key metabolite derived from dietary phytate hydrolysis and influences nutrient absorption, microbial metabolism, and gut health. Despite its physiological relevance, the identity of MI-utilizing bacteria in the chicken gastrointestinal tract remains poorly characterized. This study provides a culture-based framework to isolate and characterize anaerobically growing bacteria with potential MI-utilizing capacity from two commercial laying hen breeds, LB and LSL, across multiple intestinal regions and experimental trials.
In Trial 1, multiple gastrointestinal compartments (crop, duodenum, ileum, and caeca) were sampled because bacterial composition varies along the gut, and the primary site of MI utilization in poultry had not been determined. Based on results from Trial 1, the ileum was identified as a likely hotspot for potential MI-utilizing bacteria, and therefore, only this section was sampled in Trial 2. Although the number of animals per trial was limited, this sampling strategy was essential to capture potential spatial variation in microbial MI metabolism.
Isolates were recovered using two media formulations with distinct compositions. In Trial 1, MI served as the sole carbon source; in Trial 2, it was the principal carbon source, with soy peptone and tryptone added solely as nutritional supplements to support the growth of gut-derived bacteria with complex metabolic requirements. This modification was intended based on observations from Trial 1, where strictly minimal growth media may not have met the nutritional requirements of certain intestinal taxa, potentially contributing to the limited taxonomic diversity recovered. Although soy peptone and tryptone are not direct carbon sources, they provide peptides, amino acids, and growth factors that facilitate bacterial growth. Additionally, in both trials, media were supplemented with a defined vitamin mix and hemin to support anaerobic growth.
Growth observed under both conditions is consistent with, but does not demonstrate, MI utilization. In Trial 2, the possibility of residual or alternative carbon utilization cannot be excluded due to the presence of additional nutrients. Moreover, the initial enrichment step likely imposed selective pressure favoring fast-growing, metabolically versatile facultative anaerobes such as members of the Escherichia-Shigella clade, potentially limiting the recovery of strict or slow-growing MI-utilizing taxa. Consequently, despite nutritional supplementation, certain MI-utilizing anaerobes with specific cofactor or syntrophic requirements likely remained uncultured. The isolates recovered therefore represent only a subset of cultivable, potentially MI-utilizing gut bacteria under the applied conditions.
Using selective cultivation on MI-supplemented minimal growth media followed by partial 16S rRNA gene sequencing, 42 isolates representing ten non-redundant strains were recovered, spanning three bacterial phyla: Pseudomonadota, Bacillota, and Bacteroidota. This phylogenetic distribution aligns with previous studies reporting these phyla as dominant members of the chicken gastrointestinal communities [35,36]. Facultative anaerobes, particularly members of the Escherichia-Shigella clade, were most frequently recovered across breeds, trials, and gut sections. In contrast, taxa such as Clostridium, Enterococcus, Pediococcus, and Bacteroides were recovered sporadically and were often restricted to specific trials or breeds. The comparatively low recovery of Bacteroidota may reflect both competitive disadvantages under the selective enrichment conditions and uneven genetic distribution of IolCatGCs within this phylum [23].
The selective influence of MI-enriched cultivation is further supported by comparison with previous cultivation-based studies. Rios-Galicia et al. isolated eight anaerobic strains from the crop, gizzard, and ileum of LSL and LB laying hens and Ross 308 broilers using similar anaerobic cultivation strategies with different media formulations [37]. Their isolates were exclusively affiliated with the phylum Bacillota, represented by the genera Clostridium, Faecalispora, Ligilactobacillus, and Limosilactobacillus. In contrast, Bacillota accounted for 50% of our representative strains, but the recovered genera differed, comprising Clostridium, Enterococcus, and Pediococcus. This broader phylogenetic recovery is consistent with genome-based evidence indicating that IolCatGCs are unevenly distributed but widespread among gut-associated bacteria, underscoring the metabolic relevance of MI in shaping gut microbial communities [23].
This study also highlights limitations of 16S rRNA gene-based identification. While suitable for dereplication and broad taxonomic assignment, it does not provide reliable species-level resolution within the Enterobacteriaceae. Accordingly, Escherichia-Shigella isolates are reported at the clade level, consistent with prior reports highlighting >99% sequence similarity between these genera [33,34]. Although metabolic activity was not directly assessed, repeated recovery of Escherichia-Shigella clade isolates under selective conditions suggests metabolic flexibility or alternative survival mechanisms in MI-supplemented environments.
Breed-level observations suggest that LB hens yielded a higher absolute number of isolates, whereas LSL hens exhibited slightly greater taxonomic diversity among representative strains. However, given the limited number of animals per breed and trial, these observations should be interpreted as descriptive rather than statistically supported. Despite this limitation, the observed differences may provide preliminary insight, as samples from both breeds were collected and processed under identical experimental conditions. This pattern aligns with earlier reports on the core microbiota of LB and LSL hens, where Roth et al. reported statistically significant differences in amplicon sequence variant (ASV) profiles across five gut sections (crop, gizzard, duodenum, ileum, and caeca), with breed exerting a stronger effect than anatomical site [38]. Collectively, these observations point towards potential breed-specific colonization patterns or selective enrichment under MI-supplemented conditions, consistent with underlying host-driven differences in gut physiology and microbial ecology [38].
From a functional perspective, several genera recovered in this study have previously been reported to encode conserved inositol catabolic operons. For instance, Enterobacter sp. YB-46, isolated from soil using MI as the sole carbon source, exhibited efficient inositol degradation via the iolG- encoded MI dehydrogenase [17]. Similarly, multiple Clostridium species harbor conserved IolCatGCs, including dehydrogenase, kinase, and epimerase components [23]. Although inositol metabolism is highly strain-specific, the presence of these genera among isolates recovered under MI-enriched conditions provides preliminary, indirect evidence of potential MI utilization.
Biosafety assessment classified most isolates as RG 2, reflecting their documented opportunistic potential rather than intrinsic virulence. This aligns with a large-scale genome study showing that IolCatGCs are widely distributed among RG 2 bacteria, including many animal-associated taxa, underscoring MI metabolism as a common trait of pathogenic species [23]. However, RG classification alone does not imply pathogenicity, as virulence is highly strain-specific and cannot be inferred from 16S rRNA gene data [39]. To further address this limitation, targeted PCR screening of selected Escherichia-Shigella isolates confirmed the absence of Shigella spp., with only limited detection of the ipaH gene and no other tested virulence-associated markers. Overall, these results indicate that standard laboratory biosafety practices are sufficient, while comprehensive pathogenicity assessment would require genome-resolved analyses.
The recovery of a limited number of bacterial taxa under MI-enriched conditions may also reflect community-level metabolic interactions, in which complete MI degradation requires metabolic cross-feeding among multiple microbial species. Such syntrophic cooperation is common in complex gut ecosystems [23,40]. Supporting this concept, De Vos et al. (2024) demonstrated that Mitsuokella jalaludinii initiates phytate degradation by releasing MI intermediates that are subsequently utilized by Anaerostipes rhamnosivorans for propionate production [10]. More broadly, a recent culturomics-based study in pigs provided direct evidence of MI cross-feeding between phytase-positive and phytase-negative intestinal bacteria following phytate hydrolysis [22]. Although derived from a different host species, these findings support a model in which MI functions as a shared metabolic intermediate within intestinal microbial consortia. It is therefore plausible that MI or its derivatives in the chicken gut are similarly metabolized through consortial interactions, with distinct taxa contributing complementary enzymatic steps.
5. Conclusions
This study provides a foundational culture-based framework for isolating and characterizing bacteria with potential MI-utilizing capacity from the gastrointestinal tract of poultry. Forty-two isolates representing ten non-redundant strains across three bacterial phyla (Bacillota, Pseudomonadota, and Bacteroidota) were recovered, with facultative anaerobes, particularly members of the Escherichia-Shigella clade, predominating. Growth in MI-enriched media suggests potential MI-associated metabolic interaction; however, this evidence remains indirect and requires complementary biochemical or genomic assays such as substrate consumption profiling, enzyme activity testing, or confirmation of iol gene clusters to verify true MI catabolic activity. These findings also highlight the possibility of cooperative or complementary roles among diverse bacterial taxa in MI metabolism, though in situ activities remain to be demonstrated.
Study limitations: Several methodological constraints may have influenced the observed isolate composition. Enrichment of pooled digesta samples and peptide-rich media likely favored fast-growing facultative anaerobes, possibly underrepresenting strict anaerobes or slow-growing MI-utilizers. Species-level identification of Escherichia-Shigella isolates was limited by 16S rRNA sequence similarity. Growth on MI-enriched media alone provides indirect evidence of MI utilization, and potential syntrophic interactions within the gut were not assessed. Breed-level patterns are descriptive and not statistically supported.
Future perspectives: To comprehensively characterize MI metabolism in the chicken gut, complementary approaches combining culture-independent metagenomics, targeted multi-locus or whole-genome sequencing, and biochemical assays are recommended. Such approaches can resolve species-level identities, uncover slow-growing or strict anaerobic MI utilizers, and identify potential consortial interactions. A deeper understanding of these microbial processes could inform the development of targeted feed additives or defined probiotic formulations to enhance MI utilization, thereby improving nutrient efficiency and gut health in commercial poultry systems.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microbiolres17030046/s1, Figure S1: Gel electrophoresis showing PCR products for virulence gene detection. PCR products of selected Escherichia-Shigella isolates for virulence genes (ipaH and ial) (Pos.); Table S1: Ingredients and calculated composition of the experimental diet of the laying hens used in Trial 2; Table S2: Composition of all the media used in this study; Table S3: Number of isolates and unique RFLP patterns recovered per intestinal compartment, breed, and trial; Table S4: Detailed information of all 42 isolates, including taxonomic annotation based on NCBI, EZBioCloud, and GTDB.
Author Contributions
Conceptualization, J.S. and H.N.; methodology, H.N.; software, H.N.; validation, J.S. and H.N.; formal analysis, H.N.; investigation, H.N. and T.M.F.; resources, J.S., A.C.-S. and T.M.F.; data curation, H.N.; writing—original draft preparation, H.N.; writing—review and editing, J.S., A.C.-S., T.M.F. and H.N.; visualization, H.N.; supervision, J.S. and A.C.-S.; project administration, J.S.; funding acquisition, J.S. and A.C.-S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—Project SE 2059/7-2 within the research unit FOR 2601 (P-Fowl, project number 322916021).
Institutional Review Board Statement
The animal experiment was approved by Regierungspräsidium Tübingen (Project no. HOH67-21TE, approved on 31 January 2022) and was conducted in accordance with the German Animal Welfare Legislation.
Informed Consent Statement
Not applicable.
Data Availability Statement
The 16S rRNA gene sequences of the representative bacterial isolates generated in this study have been deposited in the European Nucleotide Archive (ENA) under accession numbers listed in Table 2 (OZ352798–OZ352797). The datasets are publicly available and can be accessed via the ENA browser at http://www.ebi.ac.uk/ena/data/view (accessed on 22 February 2026). Additional data supporting the findings of this study are available within the article and its Supplementary Materials. Other datasets generated during this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| ARDRA | Amplified Ribosomal DNA Restriction Analysis |
| ASV | Amplicon Sequence Variant |
| BLASTn | Basic Local Alignment Search Tool (nucleotide) |
| DNA | Deoxyribonucleic Acid |
| EIEC | Enteroinvasive Escherichia coli |
| ENA | European Nucleotide Archive |
| GTDB | Genome Taxonomy Database |
| IolCatGCs | Inositol catabolic gene clusters |
| LB | Lohman Brown-classic |
| LSL | Lohmann LSL-classic |
| MI | Myo-inositol |
| NCBI | National Center for Biotechnology Information |
| PBS | Phosphate-Buffered Saline |
| PCR | Polymerase Chain Reaction |
| RefSeq | Reference Sequence |
| RFLP | Restriction Fragment Length Polymorphism |
| RG | Risk Group |
| rRNA | Ribosomal Ribonucleic Acid |
| TRBA | Technical Rules for Biological Agents |
| TSB | Tryptic soy broth |
References
- Gonzalez-Uarquin, F.; Rodehutscord, M.; Huber, K. Myo-Inositol: Its Metabolism and Potential Implications for Poultry Nutrition—A Review. Poult. Sci. 2020, 99, 893–905. [Google Scholar] [CrossRef]
- Sommerfeld, V.; Hanauska, A.; Huber, K.; Bennewitz, J.; Camarinha-Silva, A.; Feger, M.; Föller, M.; Oster, M.; Ponsuksili, S.; Schmucker, S.; et al. Effects of Myo-Inositol Supplementation in the Diet on Myo-Inositol Concentrations in the Intestine, Blood, Eggs, and Excreta of Laying Hens. Poult. Sci. 2025, 104, 104545. [Google Scholar] [CrossRef]
- Lee, S.A.; Bedford, M.R. Inositol—An Effective Growth Promotor? World’s Poult. Sci. J. 2016, 72, 743–760. [Google Scholar] [CrossRef]
- Yoshida, K.; Bott, M. Microbial Synthesis of Health-Promoting Inositols. Curr. Opin. Biotechnol. 2024, 87, 103114. [Google Scholar] [CrossRef]
- Herwig, E.; Classen, H.L.; Walk, C.L.; Bedford, M.; Schwean-Lardner, K. Dietary Inositol Reduces Fearfulness and Avoidance in Laying Hens. Animals 2019, 9, 938. [Google Scholar] [CrossRef]
- Arthur, C. Understanding the Importance of Myo-Inositol for Poultry Production. Ph.D. Thesis, Harper Adams University, Newport, UK, 2024. [Google Scholar]
- Szentgyörgyi, Á.; Sommerfeld, V.; Rodehutscord, M.; Huber, K. Metabolite Profiling Under Dietary Myo-Inositol Supplementation in Laying Hens from Two High-Performing Strains. Animals 2025, 15, 1392. [Google Scholar] [CrossRef]
- Sommerfeld, V.; Huber, K.; Bennewitz, J.; Camarinha-Silva, A.; Hasselmann, M.; Ponsuksili, S.; Seifert, J.; Stefanski, V.; Wimmers, K.; Rodehutscord, M. Phytate Degradation, Myo-Inositol Release, and Utilization of Phosphorus and Calcium by Two Strains of Laying Hens in Five Production Periods. Poult. Sci. 2020, 99, 6797–6808. [Google Scholar] [CrossRef] [PubMed]
- Röhm, K.; Gonzalez-Uarquin, F.; Harmel, R.K.; Nguyen Trung, M.; Diener, M.; Fiedler, D.; Huber, K.; Seifert, J. Investigation of a Potential Electrogenic Transport-System for Myo-Inositol in the Small Intestine of Laying Hens. Br. Poult. Sci. 2022, 63, 91–97. [Google Scholar] [CrossRef] [PubMed]
- De Vos, W.M.; Nguyen Trung, M.; Davids, M.; Liu, G.; Rios-Morales, M.; Jessen, H.; Fiedler, D.; Nieuwdorp, M.; Bui, T.P.N. Phytate Metabolism Is Mediated by Microbial Cross-Feeding in the Gut Microbiota. Nat. Microbiol. 2024, 9, 1812–1827. [Google Scholar] [CrossRef]
- Henry, S.; White, M.J. Inositol-Excreting Yeast. Biotechnol. Adv. 1997, 15, 799. [Google Scholar] [CrossRef]
- Li, Y.; Han, P.; Wang, J.; Shi, T.; You, C. Production of Myo-Inositol: Recent Advance and Prospective. Biotechnol. Appl. Biochem. 2022, 69, 1101–1111. [Google Scholar] [CrossRef]
- Volk, W.A.; Pennington, D. The Fermentation of Inositol. J. Bacteriol. 1951, 61, 469–473. [Google Scholar] [CrossRef]
- Yebra, M.J.; Zúñiga, M.; Beaufils, S.; Pérez-Martínez, G.; Deutscher, J.; Monedero, V. Identification of a Gene Cluster Enabling Lactobacillus casei BL23 To Utilize Myo-Inositol. Appl. Environ. Microbiol. 2007, 73, 3850–3858. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Yamaguchi, M.; Morinaga, T.; Kinehara, M.; Ikeuchi, M.; Ashida, H.; Fujita, Y. Myo-Inositol Catabolism in Bacillus subtilis. J. Biol. Chem. 2008, 283, 10415–10424. [Google Scholar] [CrossRef] [PubMed]
- Kröger, C.; Fuchs, T.M. Characterization of the Myo-Inositol Utilization Island of Salmonella enterica Serovar Typhimurium. J. Bacteriol. 2009, 191, 545–554. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, K.-K.; Yoon, K.-H. Molecular Cloning and Characterization of myo-Inositol Dehydrogenase from Enterobacter sp. YB-46. Microbiol. Biotechnol. Lett. 2018, 46, 102–110. [Google Scholar] [CrossRef]
- Bui, T.P.N.; Mannerås-Holm, L.; Puschmann, R.; Wu, H.; Troise, A.D.; Nijsse, B.; Boeren, S.; Bäckhed, F.; Fiedler, D.; deVos, W.M. Conversion of Dietary Inositol into Propionate and Acetate by Commensal Anaerostipes Associates with Host Health. Nat. Commun. 2021, 12, 4798. [Google Scholar] [CrossRef]
- Trischler, R.; Rustler, S.M.; Poehlein, A.; Daniel, R.; Breitenbach, M.; Helfrich, E.J.N.; Müller, V. 3-Hydroxypropionate Production from myo-Inositol by the Gut Acetogen Blautia schinkii. Environ. Microbiol. 2024, 26, e16692. [Google Scholar] [CrossRef]
- Wu, C.; Yang, F.; Zhong, H.; Hong, J.; Lin, H.; Zong, M.; Ren, H.; Zhao, S.; Chen, Y.; Shi, Z.; et al. Obesity-Enriched Gut Microbe Degrades Myo-Inositol and Promotes Lipid Absorption. Cell Host Microbe 2024, 32, 1301–1314.e9. [Google Scholar] [CrossRef]
- Lee, C.-H.; Bui, T.P.N.; Petitfils, C.; Jian, C.; Wong, G.C.; Puel, A.; Roy, T.L.; Bellais, S.; Abdallah, B.B.; Nehlich, M.; et al. Novel myo-Inositol to Butyrate Fermentation Pathway in the Prevalent Human Gut Species Dysosmobacter welbionis, a Bacterium Associated with Improved Metabolic and Liver Health. Gut 2026. [Google Scholar] [CrossRef]
- Paul, L.-S.; Weber, M.; Wagner, S.; Fuchs, T.M. A Culturomics Approach Reveals Cross-Feeding Capacity of Intestinal Pig Bacteria upon Release of Inositol from Phytate. Microbiome 2026, 14, 44. [Google Scholar] [CrossRef]
- Weber, M.; Fuchs, T.M. Metabolism in the Niche: A Large-Scale Genome-Based Survey Reveals Inositol Utilization To Be Widespread among Soil, Commensal, and Pathogenic Bacteria. Microbiol. Spectr. 2022, 10, e02013-22. [Google Scholar] [CrossRef] [PubMed]
- Naithani, H.; Rios-Galicia, B.; Silva, A.C.; Seifert, J. Strategies to Enhance Cultivation of Anaerobic Bacteria from Gastrointestinal Tract of Chicken. J. Vis. Exp. (JoVE) 2024, 207, e66570. [Google Scholar] [CrossRef]
- Shahbaz, F.; Muccee, F.; Shahab, A.; Safi, S.Z.; Alomar, S.Y.; Qadeer, A. Isolation and in Vitro Assessment of Chicken Gut Microbes for Probiotic Potential. Front. Microbiol. 2024, 15, 1278439. [Google Scholar] [CrossRef] [PubMed]
- Monciardini, P.; Sosio, M.; Cavaletti, L.; Chiocchini, C.; Donadio, S. New PCR Primers for the Selective Amplification of 16S rDNA from Different Groups of Actinomycetes. FEMS Microbiol. Ecol. 2002, 42, 419–429. [Google Scholar] [CrossRef]
- Rios Galicia, B. Novel Bacterial Species from the Chicken Gastrointestinal Tract and Their Functional Diversity. Ph.D. Thesis, Universitat Hohenheim, Stuttgart, Germany, 2023. [Google Scholar]
- Kumar, S.; Stecher, G.; Suleski, M.; Sanderford, M.; Sharma, S.; Tamura, K. MEGA12: Molecular Evolutionary Genetic Analysis Version 12 for Adaptive and Green Computing. Mol. Biol. Evol. 2024, 41, msae263. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- BAuA—Regelwerk—TRBA 466 Einstufung von Prokaryonten (Bacteria Und Archaea) in Risikogruppen—Bundesanstalt Für Arbeitsschutz Und Arbeitsmedizin. Available online: https://www.baua.de/DE/Angebote/Regelwerk/TRBA/TRBA-466 (accessed on 1 September 2025).
- Bin Kingombe, C.I.; Cerqueira-Campos, M.-L.; Farber, J.M. Molecular Strategies for the Detection, Identification, and Differentiation between Enteroinvasive Escherichia coli and Shigella spp. J. Food Prot. 2005, 68, 239–245. [Google Scholar] [CrossRef]
- Thong, K.L.; Hoe, S.L.L.; Puthucheary, S.; Md Yasin, R. Detection of Virulence Genes in Malaysian Shigella species by Multiplex PCR Assay. BMC Infect Dis. 2005, 5, 8. [Google Scholar] [CrossRef]
- Liu, R.; Xu, H.; Guo, X.; Liu, S.; Qiao, J.; Ge, H.; Zheng, B.; Gou, J. Genomic Characterization of Two Escherichia fergusonii Isolates Harboring Mcr-1 Gene From Farm Environment. Front. Cell. Infect. Microbiol. 2022, 12, 774494. [Google Scholar] [CrossRef]
- Dif, G.; Djemouai, N.; Bouras, N.; Zitouni, A. In-Depth Genome-Based Analysis of Shigella spp. and Escherichia spp.: Resolving Ambiguities and Unveiling Phylogenetic Relationships. Curr. Microbiol. 2025, 82, 170. [Google Scholar] [CrossRef]
- Rychlik, I. Composition and Function of Chicken Gut Microbiota. Animals 2020, 10, 103. [Google Scholar] [CrossRef]
- Fathima, S.; Shanmugasundaram, R.; Adams, D.; Selvaraj, R.K. Gastrointestinal Microbiota and Their Manipulation for Improved Growth and Performance in Chickens. Foods 2022, 11, 1401. [Google Scholar] [CrossRef] [PubMed]
- Rios-Galicia, B.; Sáenz, J.S.; Yergaliyev, T.; Roth, C.; Camarinha-Silva, A.; Seifert, J. Novel Taxonomic and Functional Diversity of Eight Bacteria from the Upper Digestive Tract of Chicken. Int. J. Syst. Evol. Microbiol. 2024, 74, 006210. [Google Scholar] [CrossRef]
- Roth, C.; Sims, T.; Rodehutscord, M.; Seifert, J.; Camarinha-Silva, A. The Active Core Microbiota of Two High-Yielding Laying Hen Breeds Fed with Different Levels of Calcium and Phosphorus. Front. Physiol. 2022, 13, 951350. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Foster, K.R.; Comstock, L.E. The Evolution of Cooperation within the Gut Microbiota. Nature 2016, 533, 255–259. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.