Abnormal Transcytosis Mechanisms in the Pathogenesis of Hydrocephalus: A Review


Abstract
1. Introduction
1.1. Hydrocephalus
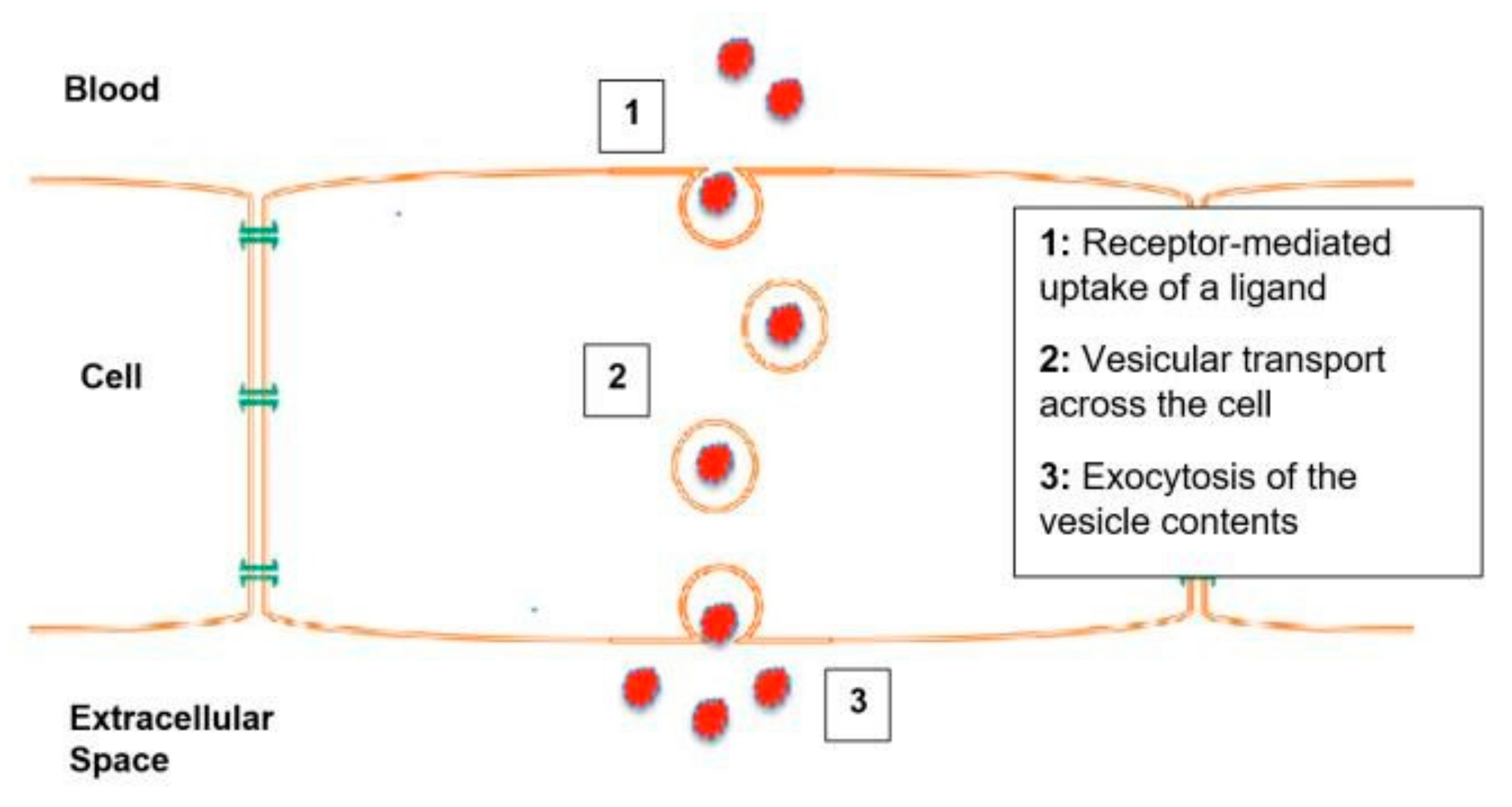
1.2. Transcytosis
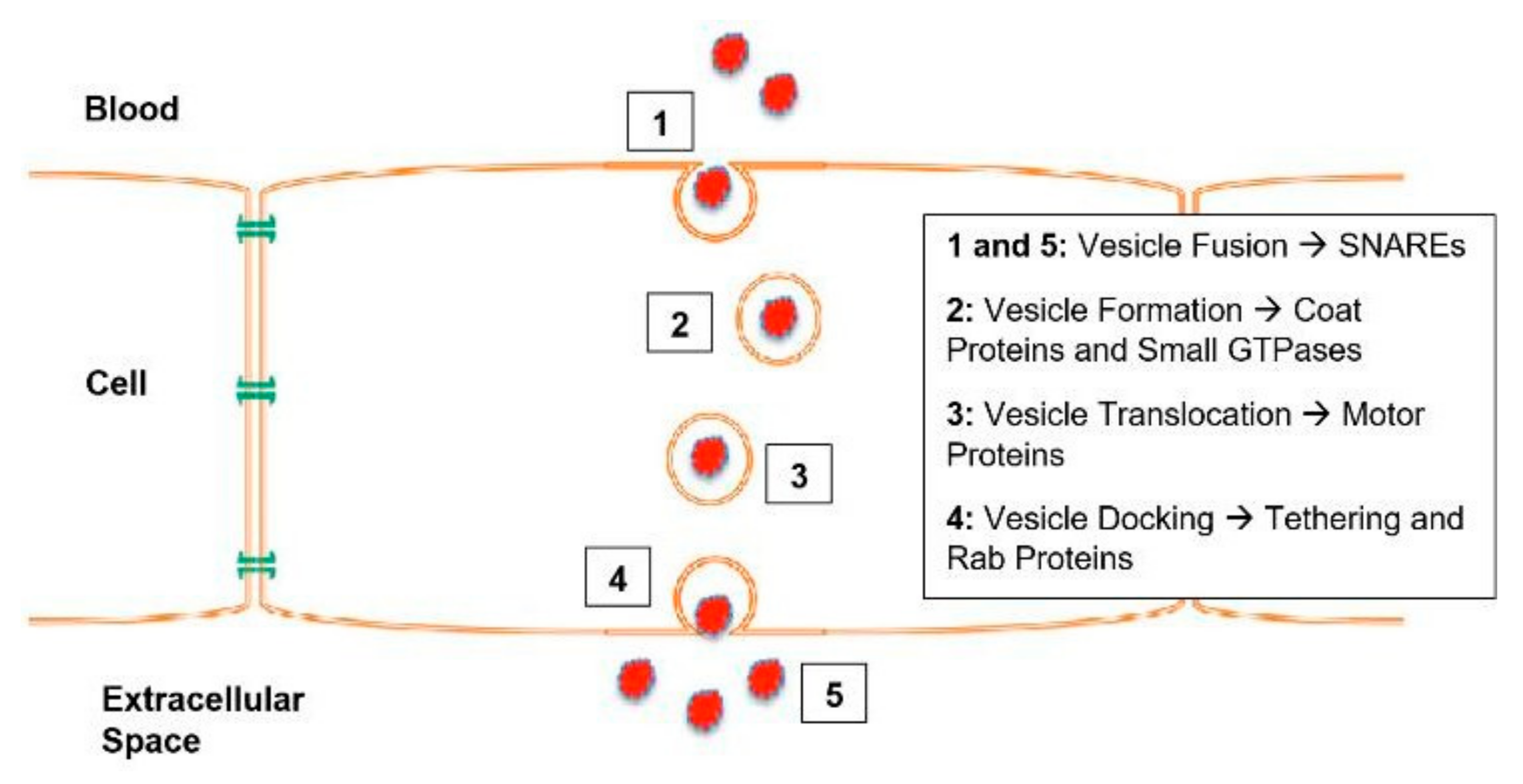
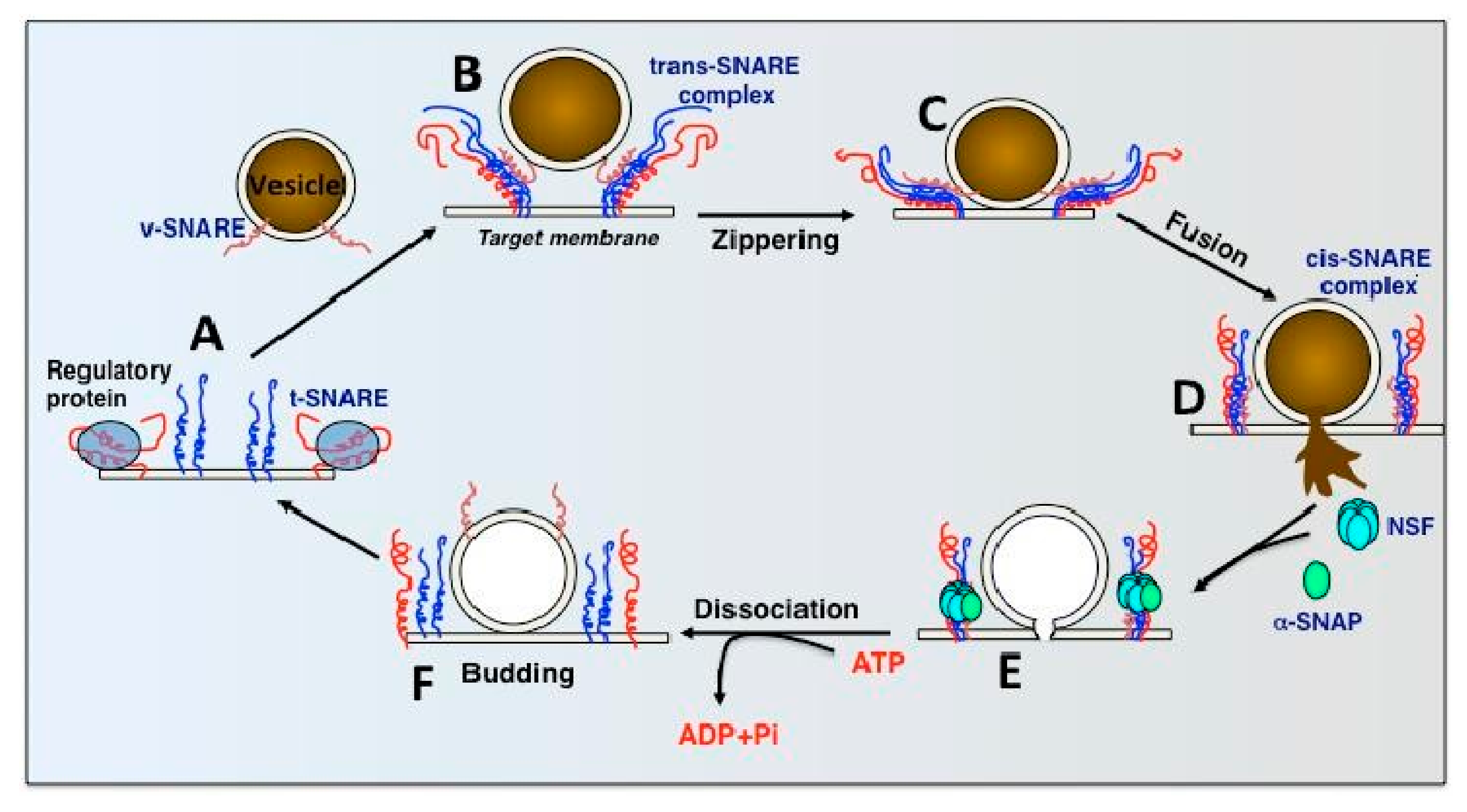
1.3. SNARE Complexes and the SNARE Hypothesis
2. N-Ethylmaleimide Sensitive Factor (NSF)
2.1. Introduction
2.2. Mechanism
2.3. NSF and Hydrocephalus
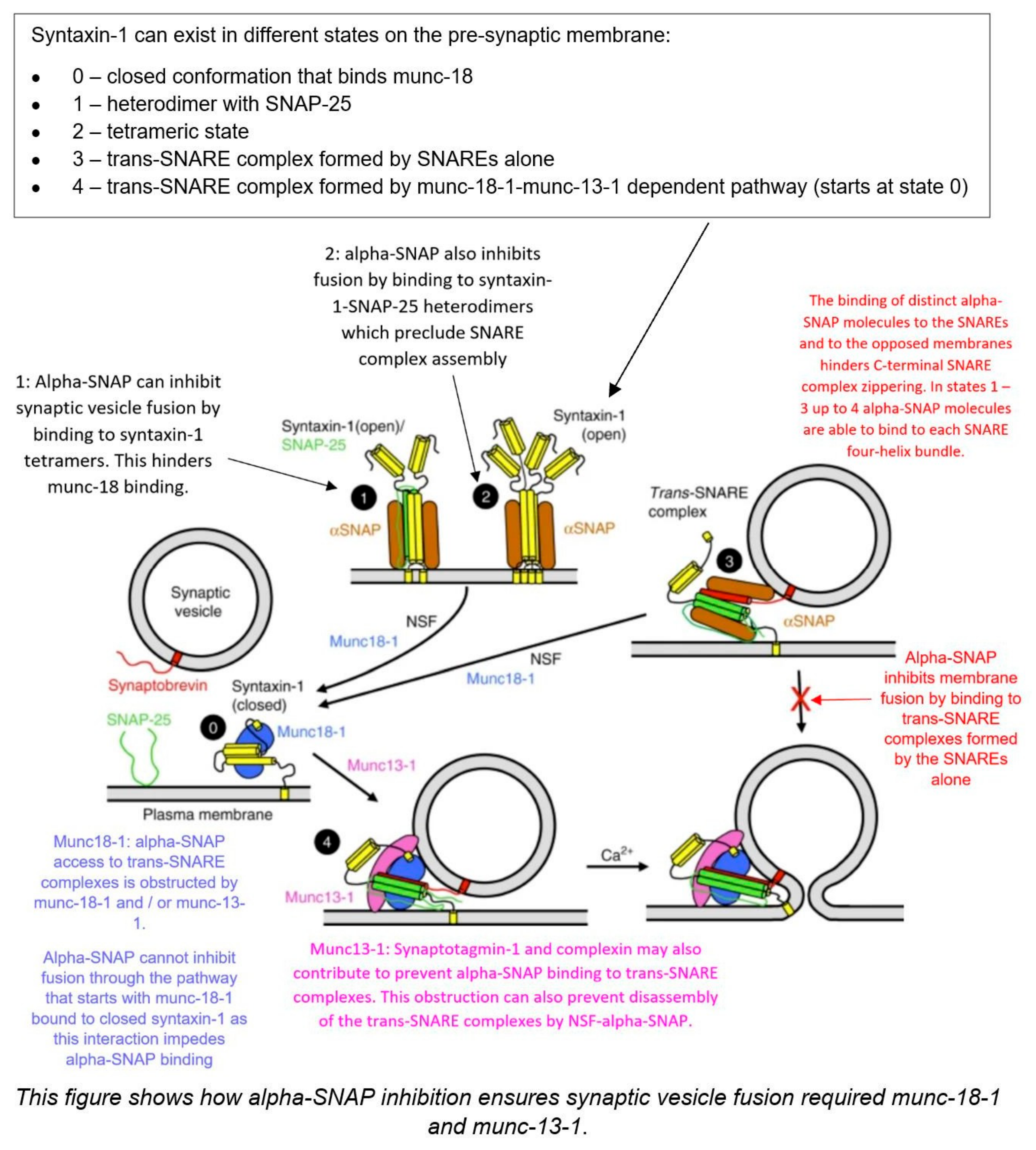
3. Alpha-SNAP
3.1. Introduction
3.2. Mechanism
3.3. Alpha-SNAP and Hydrocephalus
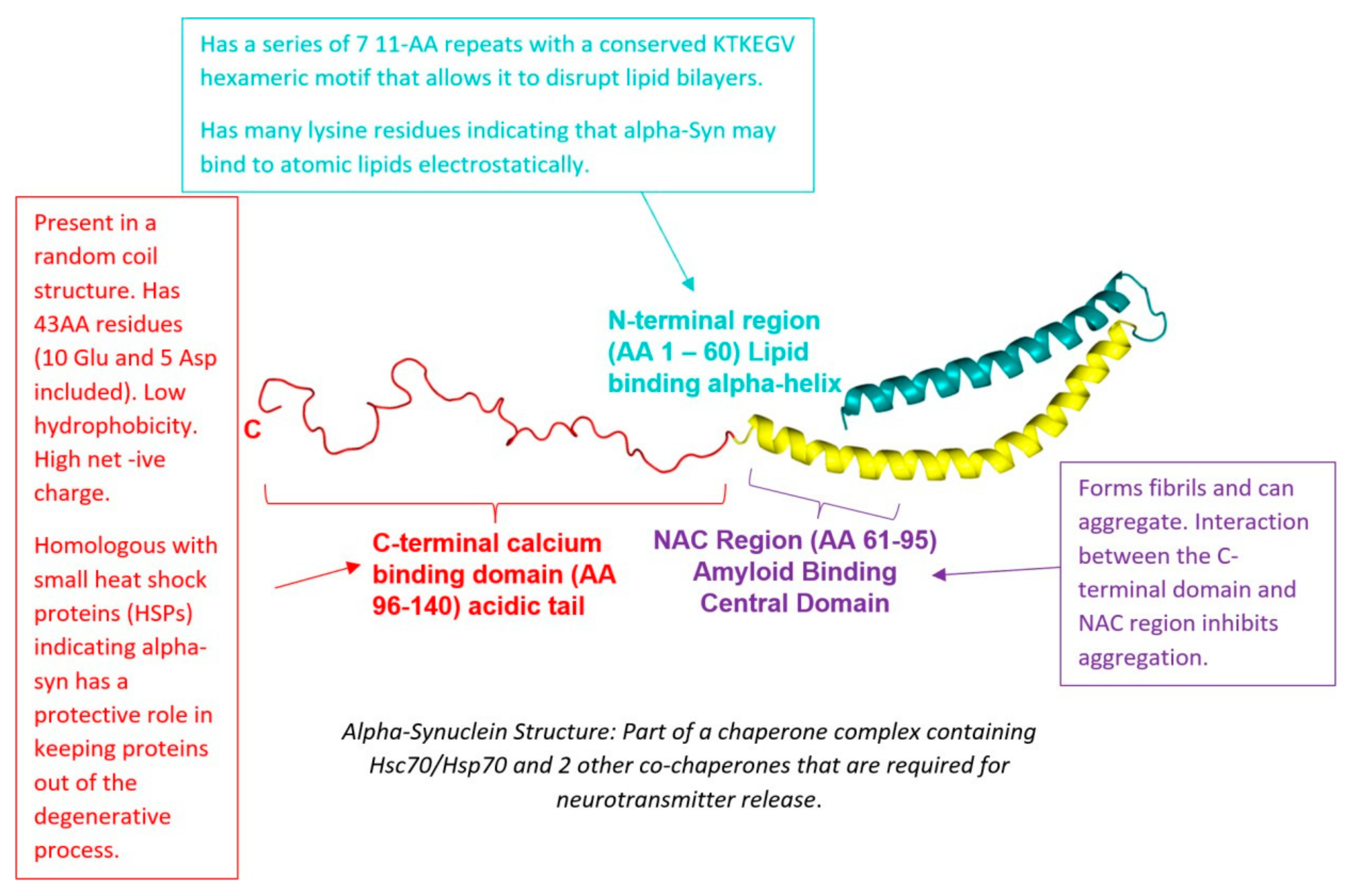
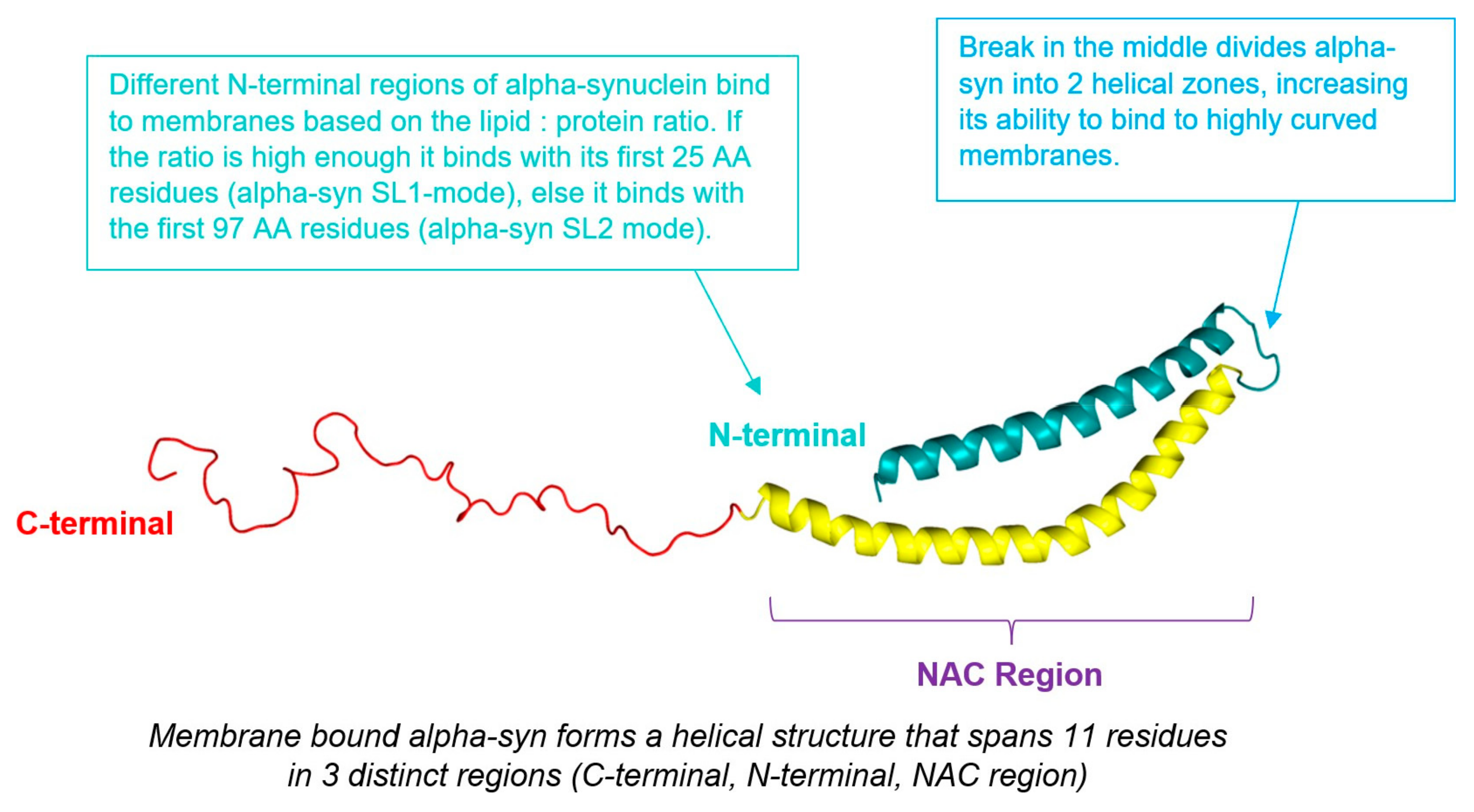
4. Alpha-Synuclein
4.1. Introduction
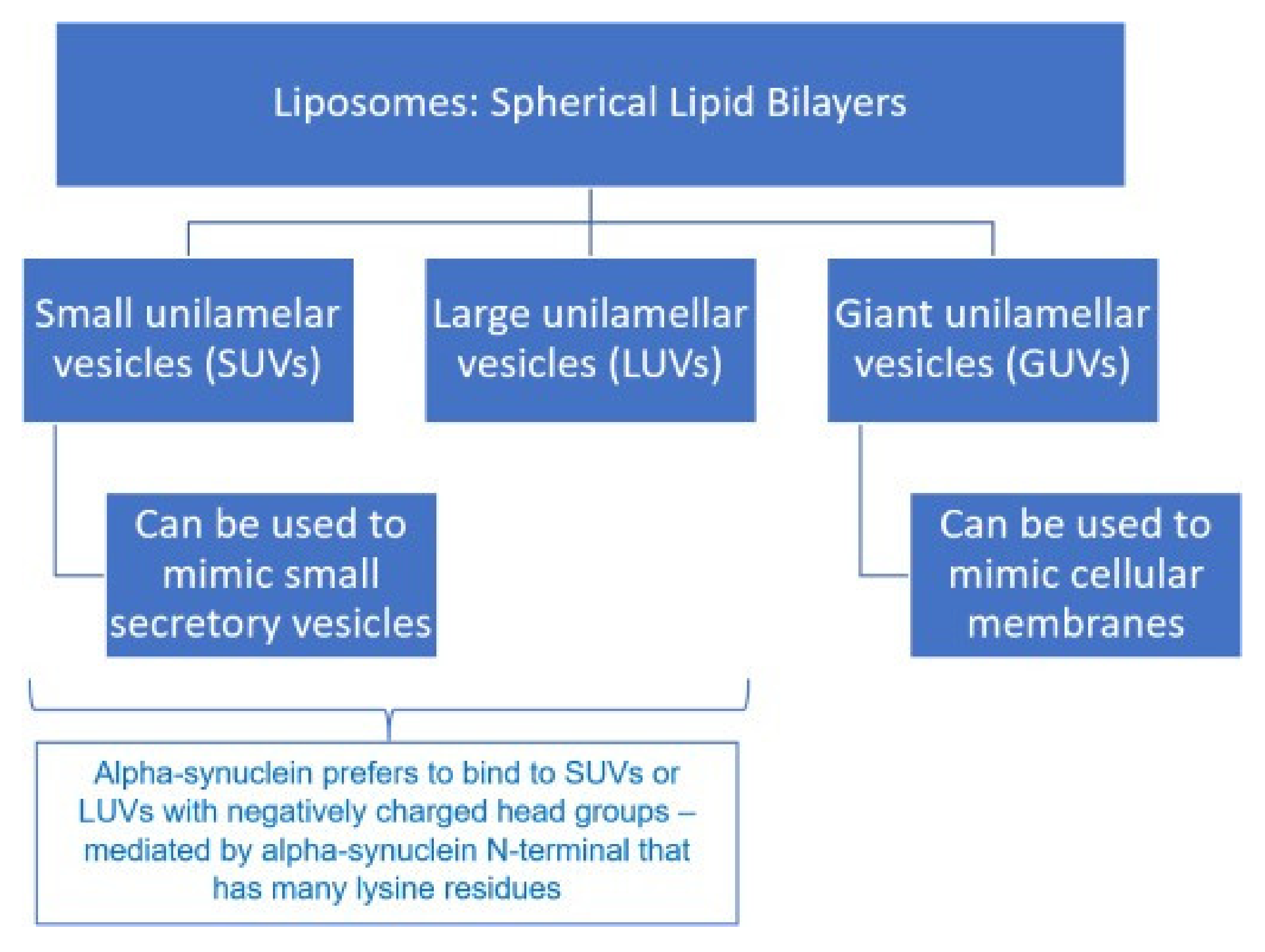
4.2. Mechanism
4.3. α-Synuclein and Hydrocephalus
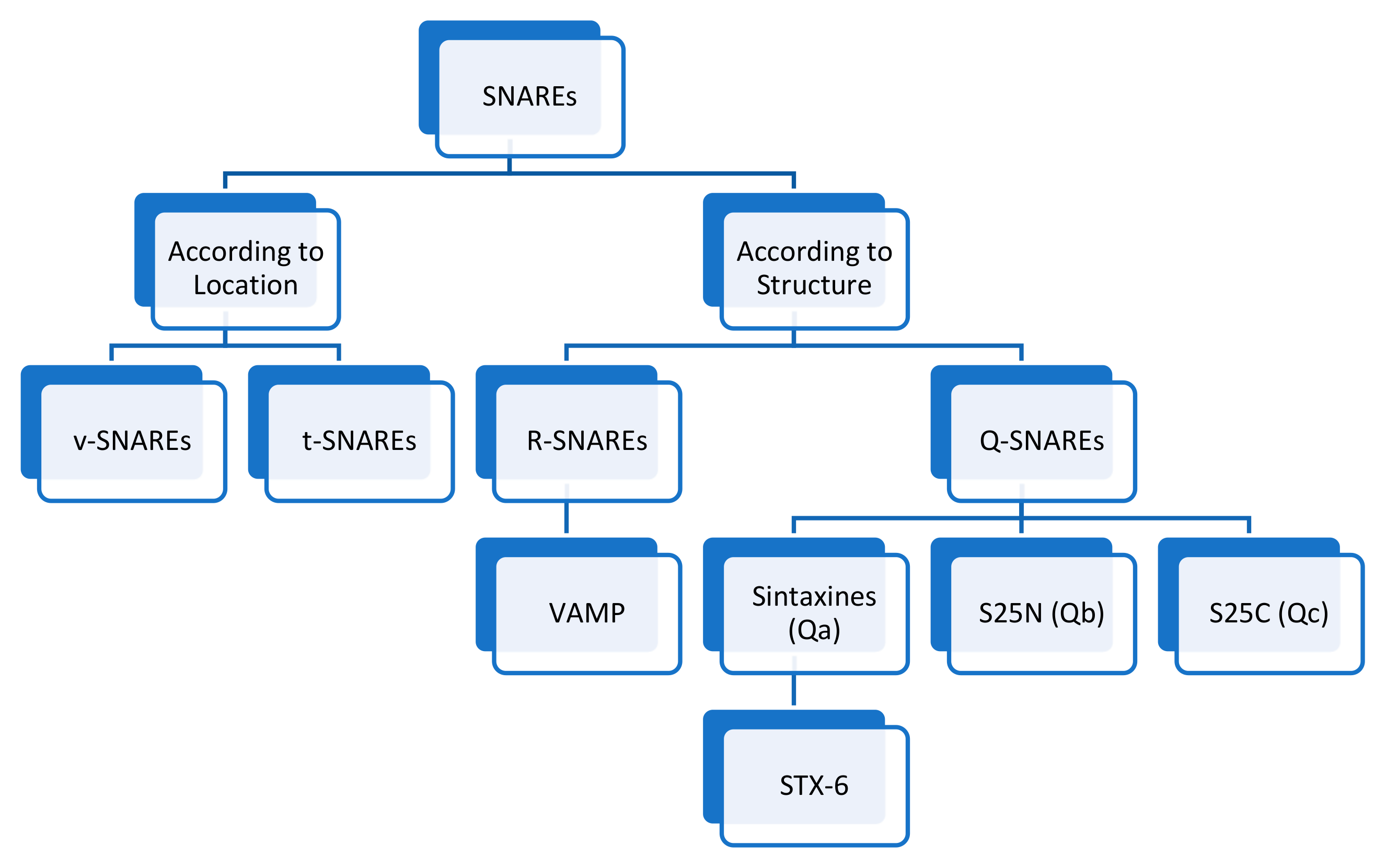
5. t-SNAREs and v-SNAREs
5.1. Introduction
5.2. Mechanism
5.3. v-SNAREs/t-SNAREs and Hydrocephalus
6. Munc18 (Sec 1) Protein
6.1. Introduction
6.2. Mechanism
6.3. Munc18 (Sec1) Protein and Hydrocephalus
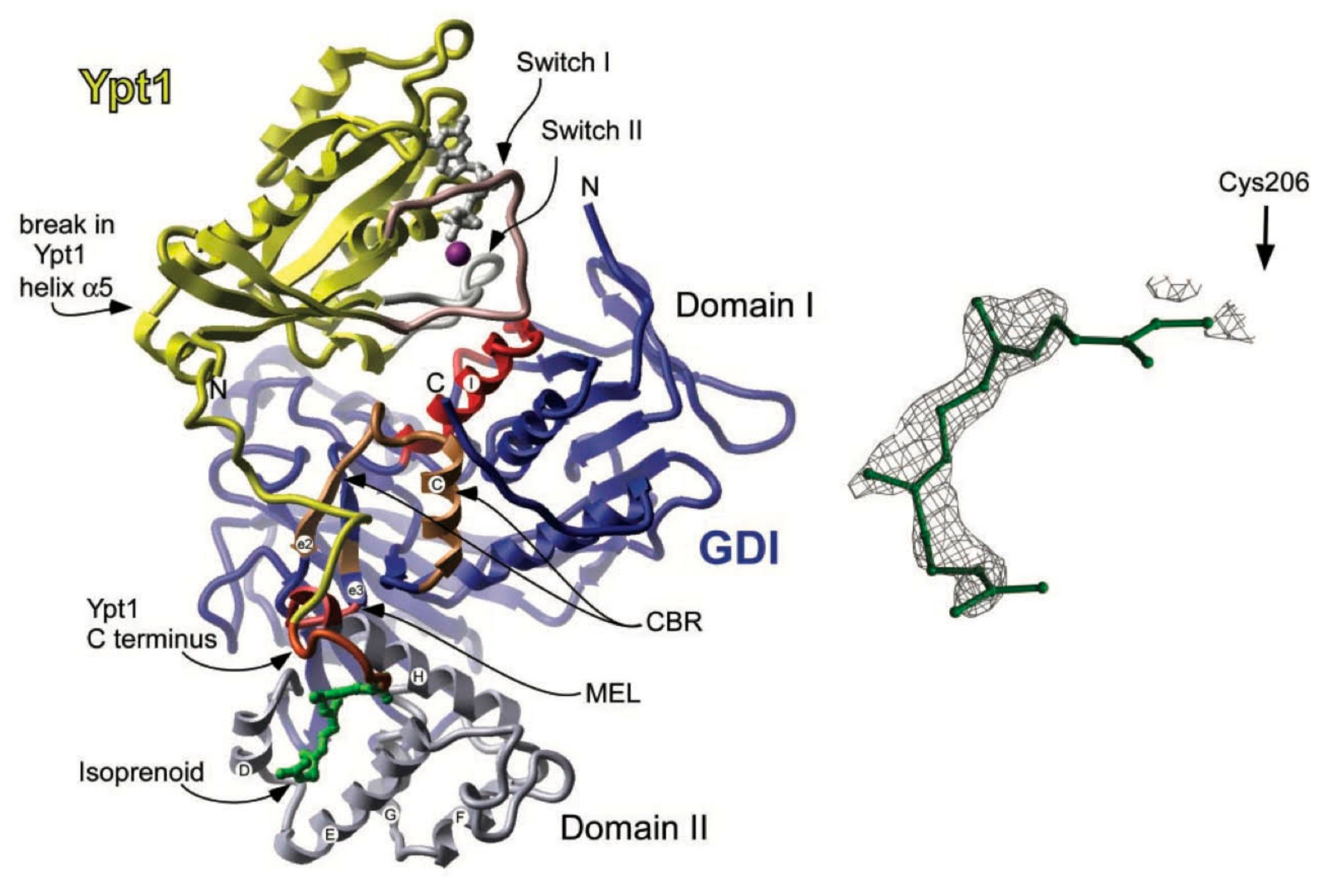
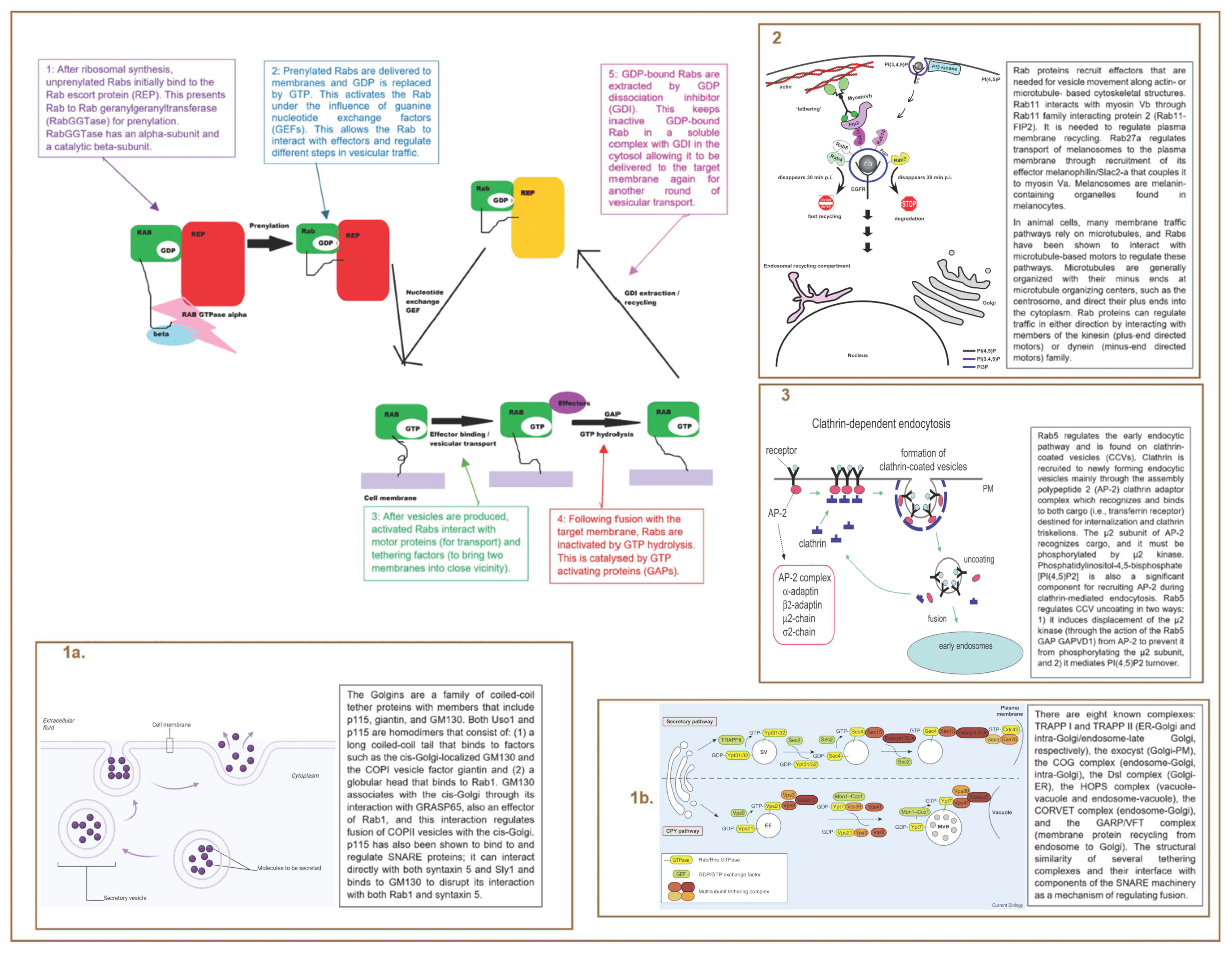
7. Rab Proteins
7.1. Introduction
7.2. Mechanism
7.3. Rab Proteins and Hydrocephalus
8. Conclusions
Funding
Conflicts of Interest
References
- Sharma, B.; Luhach, K.; Kulkarni, G.T. 4-In vitro and in vivo models of BBB to evaluate brain targeting drug delivery. In Brain Targeted Drug Delivery System; Gao, H., Gao, X., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 53–101. [Google Scholar]
- Koleva, M.; de Jesus, O. Hydrocephalus; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Bramall, A.N.; Anton, E.S.; Kahle, K.T.; Fecci, P.E. Navigating the ventricles: Novel insights into the pathogenesis of hydrocephalus. eBioMedicine 2022, 78, 103931. [Google Scholar] [CrossRef]
- Shuer, L.M. Hydrocephalus. 2024. Available online: https://www.aans.org/patients/conditions-treatments/hydrocephalus/ (accessed on 8 April 2024).
- NHS. Treatment of Hydrocephalus. 2023. Available online: https://www.nhs.uk/conditions/hydrocephalus/treatment/ (accessed on 14 February 2025).
- Agre, P. Nobel Lecture. Aquaporin water channels. Biosci. Rep. 2004, 24, 127–163. [Google Scholar] [CrossRef] [PubMed]
- Maraković, J.; Oresković, D.; Rados, M.; Vukić, M.; Jurjević, I.; Chudy, D.; Klarica, M. Effect of osmolarity on CSF volume during ventriculo-aqueductal and ventriculo-cisternal perfusions in cats. Neurosci. Lett. 2010, 484, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Orešković, D.; Radoš, M.; Klarica, M. New Concepts of Cerebrospinal Fluid Physiology and Development of Hydrocephalus. Pediatr. Neurosurg. 2017, 52, 417–425. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Li, J.; Shen, Y.; Duncan, T.M.; Jenrow, K.A.; Haacke, E.M. Normal macromolecular clearance out of the ventricles is delayed in hydrocephalus. Brain Res. 2018, 1678, 337–355. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, S.; Li, J.; Schultz, L.; Jenrow, K.A. Increased CSF osmolarity reversibly induces hydrocephalus in the normal rat brain. Fluids Barriers CNS 2012, 9, 13. [Google Scholar] [CrossRef]
- College, D. Transcytosis. 2023. Available online: https://www.davidson.edu/academic-departments/biology (accessed on 14 February 2025).
- Whiteheart, S.W.; Schraw, T.; Matveeva, E.A. N-ethylmaleimide sensitive factor (NSF) structure and function. Int. Rev. Cytol. 2001, 207, 71–112. [Google Scholar]
- Vink, R.; Gabrielian, L.; Thornton, E. The Role of Substance P in Secondary Pathophysiology after Traumatic Brain Injury. Front. Neurol. 2017, 8, 304. [Google Scholar] [CrossRef]
- Wang, T.; Li, L.; Hong, W. SNARE proteins in membrane trafficking. Traffic 2017, 18, 767–775. [Google Scholar] [CrossRef]
- Marz, K.E.; Lauer, J.M.; Hanson, P.I. Defining the SNARE complex binding surface of alpha-SNAP: Implications for SNARE complex disassembly. J. Biol. Chem. 2003, 278, 27000–27008. [Google Scholar] [CrossRef]
- Ryu, J.K.; Jahn, R.; Yoon, T.Y. Review: Progresses in understanding N-ethylmaleimide sensitive factor (NSF) mediated disassembly of SNARE complexes. Biopolymers 2016, 105, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Goody, R.S.; Müller, M.P.; Wu, Y.W. Mechanisms of action of Rab proteins, key regulators of intracellular vesicular transport. Biol. Chem. 2017, 398, 565–575. [Google Scholar] [CrossRef] [PubMed]
- May, A.P.; Whiteheart, S.W.; Weis, W.I. Unraveling the Mechanism of the Vesicle Transport ATPase NSF, the N-Ethylmaleimide-sensitive Factor. J. Biol. Chem. 2001, 276, 21991–21994. [Google Scholar] [CrossRef]
- Ma, L.; Kang, Y.; Jiao, J.; Rebane, A.A.; Cha, H.K.; Xi, Z.; Qu, H.; Zhang, Y. α-SNAP Enhances SNARE Zippering by Stabilizing the SNARE Four-Helix Bundle. Cell Rep. 2016, 15, 531–539. [Google Scholar] [CrossRef]
- Huang, X.; Sun, S.; Wang, X.; Fan, F.; Zhou, Q.; Lu, S.; Cao, Y.; Wang, Q.W.; Dong, M.Q.; Yao, J.; et al. Mechanistic insights into the SNARE complex disassembly. Sci. Adv. 2019, 5, eaau8164. [Google Scholar] [CrossRef]
- Emamzadeh, F.N. Alpha-synuclein structure, functions, and interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef]
- Chua, C.E.; Tang, B.L. Rabs, SNAREs and α-synuclein--membrane trafficking defects in synucleinopathies. Brain Res. Rev. 2011, 67, 268–281. [Google Scholar] [CrossRef]
- Udayangani, S. What is the Difference Between v-SNARE and t-SNARE. 2022. Available online: https://www.differencebetween.com/what-is-the-difference-between-v-snare-and-t-snare/ (accessed on 9 September 2023).
- Goda, Y. SNAREs and regulated vesicle exocytosis. Proc. Natl. Acad. Sci. USA 1997, 94, 769–772. [Google Scholar] [CrossRef]
- Lou, X.; Shin, Y.K. SNARE zippering. Biosci. Rep. 2016, 36, e00327. [Google Scholar] [CrossRef]
- Toonen, R.F.G.; Verhage, M. Munc18-1 in secretion: Lonely Munc joins SNARE team and takes control. Trends Neurosci. 2007, 30, 564–572. [Google Scholar] [CrossRef]
- Stepien, K.P.; Prinslow, E.A.; Rizo, J. Munc18-1 is crucial to overcome the inhibition of synaptic vesicle fusion by αSNAP. Nat. Commun. 2019, 10, 4326. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef]
- Lechuga, S.; Naydenov, N.G.; Feygin, A.; Jimenez, A.J.; Ivanov, A.I. A vesicle trafficking protein αSNAP regulates Paneth cell differentiation in vivo. Biochem. Biophys. Res. Commun. 2017, 486, 951–957. [Google Scholar] [CrossRef]
- Ferland, R.J.; Batiz, L.F.; Neal, J.; Lian, G.; Bundock, E.; Lu, J.; Hsiao, Y.C.; Diamond, R.; Mei, D.; Banham, A.H.; et al. Disruption of neural progenitors along the ventricular and subventricular zones in periventricular heterotopia. Hum. Mol. Genet. 2009, 18, 497–516. [Google Scholar] [CrossRef]
- Cui-Cui, L.; Shan, S.; Sen-Fang, S. The Role of the N-D1 Linker of the N-Ethylmaleimide-Sensitive Factor in the SNARE Disassembly. PLoS ONE 2013, 8, e64346. [Google Scholar]
- Hong, H.K.; Chakravarti, A.; Takahashi, J.S. The gene for soluble N-ethylmaleimide sensitive factor attachment protein alpha is mutated in hydrocephaly with hop gait (hyh) mice. Proc. Natl. Acad. Sci. USA 2004, 101, 1748–1753. [Google Scholar] [CrossRef]
- Bátiz, L.F.; Páez, P.; Jiménez, A.J.; Rodríguez, S.; Wagner, C.; Pérez-Fígares, J.M.; Rodríguez, E.M. Heterogeneous expression of hydrocephalic phenotype in the hyh mice carrying a point mutation in alpha-SNAP. Neurobiol. Dis. 2006, 23, 152–168. [Google Scholar] [CrossRef]
- Bajjalieh, S. Trafficking in cell fate. Nat. Genet. 2004, 36, 216–217. [Google Scholar] [CrossRef]
- Chae, T.H.; Kim, S.; Marz, K.E.; Hanson, P.I.; Walsh, C.A. The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat. Genet. 2004, 36, 264–270. [Google Scholar] [CrossRef]
- Miao, Y.; Bhushan, J.; Dani, A.; Vig, M. Na+ influx via Orai1 inhibits intracellular ATP-induced mTORC2 signaling to disrupt CD4 T cell gene expression and differentiation. eLife 2017, 6, e25155. [Google Scholar] [CrossRef]
- Wang, L.; Brautigan, D.L. α-SNAP inhibits AMPK signaling to reduce mitochondrial biogenesis and dephosphorylates Thr172 in AMPKα in vitro. Nat. Commun. 2013, 4, 1559. [Google Scholar] [CrossRef] [PubMed]
- Bátiz, L.F.; De Blas, G.A.; Michaut, M.A.; Ramírez, A.R.; Rodríguez, F.; Ratto, M.H.; Oliver, C.; Tomes, C.N.; Rodríguez, E.M.; Mayorga, L.S. Sperm from hyh mice carrying a point mutation in alphaSNAP have a defect in acrosome reaction. PLoS ONE 2009, 4, e4963. [Google Scholar] [CrossRef]
- Bátiz, L.F.; Roales-Buján, R.; Rodríguez-Pérez, L.M.; Matas, I.M.; Páez, P.; Roque, M.; Jiménez, A.J.; Ramos, C.; Pérez-Fígares, J.M. A simple PCR-based genotyping method for M105I mutation of alpha-SNAP enhances the study of early pathological changes in hyh phenotype. Mol. Cell Probes 2009, 23, 281–290. [Google Scholar] [CrossRef]
- de Paola, M.; Miró, M.P.; Ratto, M.; Bátiz, L.F.; Michaut, M.A. Pleiotropic effects of alpha-SNAP M105I mutation on oocyte biology: Ultrastructural and cellular changes that adversely affect female fertility in mice. Sci. Rep. 2019, 9, 17374. [Google Scholar] [CrossRef]
- SinoBiological. Alpha SNAP. 2022. Available online: https://jp.sinobiological.com/resource/alpha-snap/proteins (accessed on 14 February 2025).
- UniProt. P37840 SYUA_HUMAN. 2023. Available online: https://www.uniprot.org/uniprotkb/P37840/entry#function (accessed on 14 February 2025).
- Vanderstichele, H.; Demeyer, L.; Janelidze, S.; Coart, E.; Stoops, E.; Mauroo, K.; Herbst, V.; François, C.; Hansson, O. Recommendations for cerebrospinal fluid collection for the analysis by ELISA of neurogranin trunc P75, α-synuclein, and total tau in combination with Aβ(1-42)/Aβ(1-40). Alzheimers Res. Ther. 2017, 9, 40. [Google Scholar] [CrossRef]
- Alpha-synuclein. 2025. Available online: https://en.wikipedia.org/w/index.php?title=Alpha-synuclein&oldid=1267677851 (accessed on 14 February 2025).
- Sakurai, A.; Tsunemi, T.; Ishiguro, Y.; Okuzumi, A.; Hatano, T.; Hattori, N. Comorbid alpha synucleinopathies in idiopathic normal pressure hydrocephalus. J. Neurol. 2022, 269, 2022–2029. [Google Scholar] [CrossRef]
- Hoshi, K.; Matsumoto, Y.; Ito, H.; Saito, K.; Honda, T.; Yamaguchi, Y.; Hashimoto, Y. A unique glycan-isoform of transferrin in cerebrospinal fluid: A potential diagnostic marker for neurological diseases. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2473–2478. [Google Scholar] [CrossRef]
- Mehta, N.H.; Suss, R.A.; Dyke, J.P.; Theise, N.D.; Chiang, G.C.; Strauss, S.; Saint-Louis, L.; Li, Y.; Pahlajani, S.; Babaria, V.; et al. Quantifying cerebrospinal fluid dynamics: A review of human neuroimaging contributions to CSF physiology and neurodegenerative disease. Neurobiol. Dis. 2022, 170, 105776. [Google Scholar] [CrossRef]
- Jurjević, I.; Miyajima, M.; Ogino, I.; Akiba, C.; Nakajima, M.; Kondo, A.; Kikkawa, M.; Kanai, M.; Hattori, N.; Arai, H. Decreased Expression of hsa-miR-4274 in Cerebrospinal Fluid of Normal Pressure Hydrocephalus Mimics with Parkinsonian Syndromes. J. Alzheimers Dis. 2017, 56, 317–325. [Google Scholar] [CrossRef]
- Connell, E.; Darios, F.; Broersen, K.; Gatsby, N.; Peak-Chew, S.Y.; Rickman, C.; Davletov, B. Mechanism of arachidonic acid action on syntaxin-Munc18. EMBO Rep. 2007, 8, 414–419. [Google Scholar] [CrossRef]
- Söllner, T.H. A mutation in the general membrane trafficking machinery and hydrocephaly. Proc. Natl. Acad. Sci. USA 2004, 101, 1431–1432. [Google Scholar] [CrossRef] [PubMed]
- Nechiporuk, T.; Fernandez, T.E.; Vasioukhin, V. Failure of epithelial tube maintenance causes hydrocephalus and renal cysts in Dlg5−/− mice. Dev. Cell 2007, 13, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Rak, A.; Pylypenko, O.; Durek, T.; Watzke, A.; Kushnir, S.; Brunsveld, L.; Waldmann, H.; Goody, R.S.; Alexandrov, K. Structure of Rab GDP-dissociation inhibitor in complex with prenylated YPT1 GTPase. Science 2003, 302, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; He, M.; Port, S.A.; Baker, R.W.; Xu, Y.; Qu, H.; Xiong, Y.; Wang, Y.; Jin, H.; Eisemann, T.J.; et al. Munc18-1 catalyzes neuronal SNARE assembly by templating SNARE association. eLife 2018, 7, e41771. [Google Scholar] [CrossRef]
- Corbeel, L.; Freson, K. Rab proteins and Rab-associated proteins: Major actors in the mechanism of protein-trafficking disorders. Eur. J. Pediatr. 2008, 167, 723–729. [Google Scholar] [CrossRef]
- Mölleken, K.; Hegemann, J.H. Acquisition of Rab11 and Rab11-Fip2-A novel strategy for Chlamydia pneumoniae early survival. PLoS Pathog. 2017, 13, e1006556. [Google Scholar] [CrossRef]
- López-Hernández, T.; Haucke, V.; Maritzen, T. Endocytosis in the adaptation to cellular stress. Cell Stress. 2020, 4, 230–247. [Google Scholar] [CrossRef]
- Witkos, T.M.; Lowe, M. Recognition and tethering of transport vesicles at the Golgi apparatus. Curr. Opin. Cell Biol. 2017, 47, 16–23. [Google Scholar] [CrossRef]
- Bröcker, C.; Engelbrecht-Vandré, S.; Ungermann, C. Multisubunit tethering complexes and their role in membrane fusion. Curr. Biol. 2010, 20, R943–R952. [Google Scholar] [CrossRef]
- McCaffrey, M.W.; Lindsay, A.J. Rab Family. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 1–6. [Google Scholar]
- Ding, X.; Fragoza, R.; Singh, P.; Zhang, S.; Yu, H.; Schimenti, J.C. Variants in RABL2A causing male infertility and ciliopathy. Hum. Mol. Genet. 2020, 29, 3402–3411. [Google Scholar] [CrossRef]
- Satir, P.; Christensen, S.T. Overview of structure and function of mammalian cilia. Annu. Rev. Physiol. 2007, 69, 377–400. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, L.B.; Rosenbaum, J.L. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr. Top. Dev. Biol. 2008, 85, 23–61. [Google Scholar] [PubMed]
- Nachury, M.V.; Seeley, E.S.; Jin, H. Trafficking to the ciliary membrane: How to get across the periciliary diffusion barrier? Annu. Rev. Cell Dev. Biol. 2010, 26, 59–87. [Google Scholar] [CrossRef]
- Moseley-Alldredge, M.; Sheoran, S.; Yoo, H.; O’Keefe, C.; Richmond, J.E.; Chen, L. A role for the Erk MAPK pathway in modulating SAX-7/L1CAM-dependent locomotion in Caenorhabditis elegans. Genetics 2022, 220, iyab215. [Google Scholar] [CrossRef]
- Rajadhyax, M.; Neti, G.; Crow, Y.; Tyagi, A. Neurological presentation of Griscelli syndrome: Obstructive hydrocephalus without haematological abnormalities or organomegaly. Brain Dev. 2007, 29, 247–250. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Caveolae Mediated | Clathrin Mediated |
|---|---|
| Make us of caveolae | Make use of clathrin protein |
| Caveolae are distinguished by the presence of caveolins and are pits in the apical and basal membranes of all endothelial cells | Clathrin is located on both apical and basal surfaces of epithelial cells and line these vesicles. On the surfcae of the cell a pit forms from specific cell receptors that are coated by clathrin |
| Mostly responsible for transcellular translocation of macromolecules in epithelal cells | Used by epithelial cells to sort through the molecules entering the cell as one of the destinations of these vesicles is the Golgi. Vesciles attach to the endoplasmic reticulum before being sorted to either the apical or basal side of the cell |
| Transport cargi, usually fluid, through the cells | Required for immune responses |
| Caveolae can merge to create arrangements including a tunnel or channel, to move cargo through the cell | Clathrin stabilizes the forming vescile by forming a rigid matrix of an assembling of clathrin proteins, which can later disassemble after the vesicle has disassociated from the membrane |
Molecules chaperone and assists in the folding of SNAREs at pre-synaptic plasma membranes alongside cysteine string protein-alphs/DNAJC5
|
| Regulate dopamine neurotransmission and modulates its activity via the dopamine transporter (DAT1) |
| Neuronal protein that regulates synaptic vesicle trafficing and neurotransmitter release |
| Promotes vesicle priming, fusion and dilation of exocytotic fusion pores |
Increases local Ca2+ release from microdomains
|
| v-SNARES | t-SNARES |
|---|---|
| Found on membrane transport vesicle during the budding process of exocytosis (usually incorporated into the membrane of transport vesicles) | Associated with nerve terminal membranes |
| e.g., VAMP7 and VAMP8 | e.g., Syntaxin 1 and SNAP-25 |
| Have more than 70% of branced amino acids in the transmembrane region | Form stable subcomplexes and function as a guide for v-SNAREs |
| Aid exocytosis of large zymogen granules and mast cell vesicles | Specific localization to subcellular membranes defines where transport vesicles bind and fuse |
| Facilitate rapid pore expansion and release of bulky macromolecules such as interferons |
| Regulate protein transport in endocytic and exocytic pathways |
| Participate in vesicle budding, membrane fusion and cytoskeletal interactions |
| Act as key regulators in intracellular vesicular transport |
Regulate pathways by interacting with effector proteins:
|
| Introduction | Mechanism | Link to Hydrocephalus | |
|---|---|---|---|
| NSF |
|
|
|
| Alpha-SNAP |
|
|
|
| Alpha-Synuclein |
| SNARE Complex Assembly:
|
|
| t-SNARES and v-SNARES |
|
|
|
| Mun18 (Sec1) Proteins |
|
|
|
| Rab Proteins | Molecular Switches:
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Randeni, A.; Colvin, S.; Krishnamurthy, S. Abnormal Transcytosis Mechanisms in the Pathogenesis of Hydrocephalus: A Review. Int. J. Mol. Sci. 2025, 26, 4881. https://doi.org/10.3390/ijms26104881
Randeni A, Colvin S, Krishnamurthy S. Abnormal Transcytosis Mechanisms in the Pathogenesis of Hydrocephalus: A Review. International Journal of Molecular Sciences. 2025; 26(10):4881. https://doi.org/10.3390/ijms26104881
Chicago/Turabian StyleRandeni, Adithi, Sydney Colvin, and Satish Krishnamurthy. 2025. "Abnormal Transcytosis Mechanisms in the Pathogenesis of Hydrocephalus: A Review" International Journal of Molecular Sciences 26, no. 10: 4881. https://doi.org/10.3390/ijms26104881
APA StyleRandeni, A., Colvin, S., & Krishnamurthy, S. (2025). Abnormal Transcytosis Mechanisms in the Pathogenesis of Hydrocephalus: A Review. International Journal of Molecular Sciences, 26(10), 4881. https://doi.org/10.3390/ijms26104881