
Melanoma Glycome Regulates the Pro-Oncogenic Properties of Extracellular Galectin-3


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
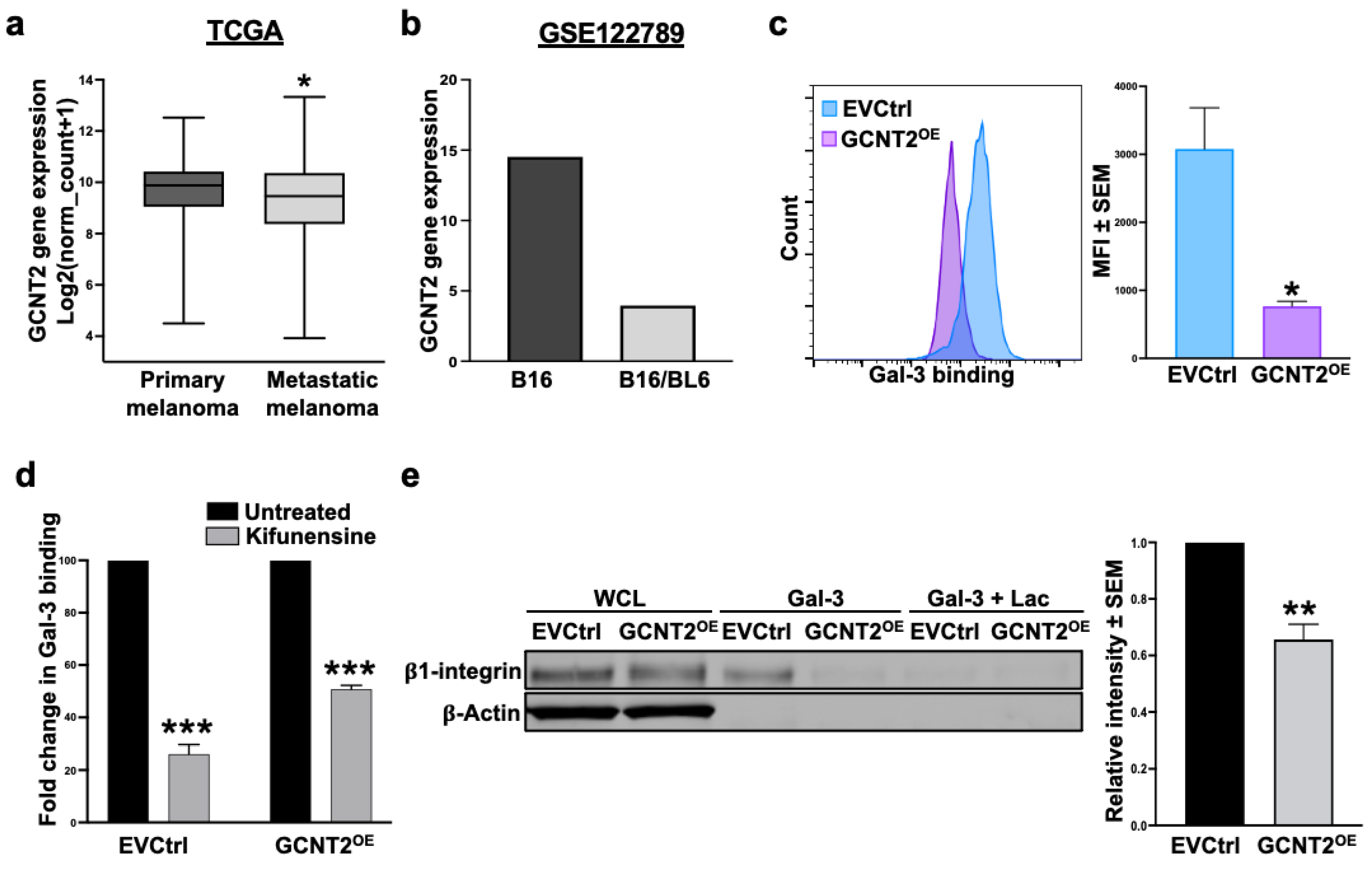
2.1. GCNT2 Is Downregulated in Human and Murine Metastatic Melanomas Compared to Primary Melanomas
2.2. I-Branching Hinders Gal-3 Binding to Its Ligands on the Melanoma Cell Surface
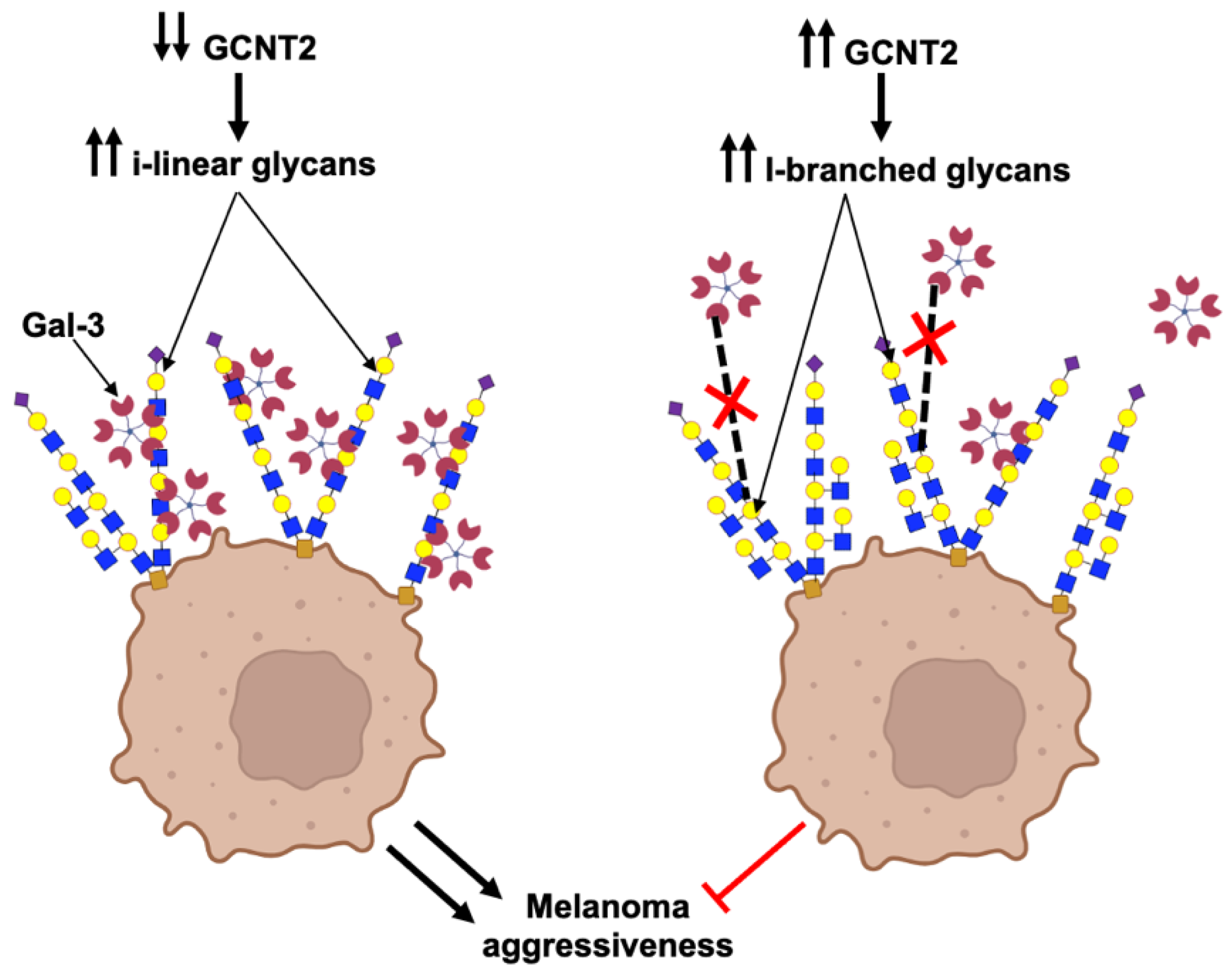
2.3. I-Branching Attenuates Gal-3-Dependent Malignant Characteristics of Melanoma Cells
3. Discussion
4. Materials and Methods
4.1. Gene Expression Data Collection and Processing
4.2. Cell Lines and Cell Culture
4.3. Cell Proliferation Assay
4.4. Transwell Migration Assay
4.5. RT-qPCR Analysis
4.6. Immunoblotting
4.7. Gal-3 Affinity Chromatography
4.8. Flow Cytometry
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef]
- Chan, P.Y.; Corrie, P.G. Curing stage IV melanoma: Where have we been and where are we? Am. Soc. Clin. Oncol. Educ. Book. 2024, 44, e438654. [Google Scholar] [CrossRef]
- Dimitrova, M.; Weber, J. Melanoma—Modern Treatment for Metastatic Melanoma. Cancer J. 2024, 30, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Vandyck, H.H.; Hillen, L.M.; Bosisio, F.M.; van den Oord, J.; Zur Hausen, A.; Winnepenninckx, V. Rethinking the biology of metastatic melanoma: A holistic approach. Cancer Metastasis Rev. 2021, 40, 603–624. [Google Scholar] [CrossRef] [PubMed]
- Niveau, C.; Sosa Cuevas, E.; Saas, P.; Aspord, C. Glycans in melanoma: Drivers of tumour progression but sweet targets to exploit for immunotherapy. Immunology 2024, 173, 33–52. [Google Scholar] [CrossRef]
- Nandi, S.; Ghosh, S.; Ranjan, A.; Sood, R.S.; Pal, J.K.; Hajela, K.; Gupta, R.K. Lectins in Health and Diseases: Galectins and Cancer. In Lectins: Innate Immune Defense and Therapeutics; Springer: Berlin/Heidelberg, Germany, 2022; pp. 215–271. [Google Scholar]
- De Vellis, C.; Pietrobono, S.; Stecca, B. The role of glycosylation in melanoma progression. Cells 2021, 10, 2136. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, J.G.; Liang, J.; Antonopoulos, A.; Giovannone, N.; Kang, S.; Mondala, T.S.; Head, S.R.; King, S.L.; Tani, Y.; Brackett, D. Loss of GCNT2/I-branched glycans enhances melanoma growth and survival. Nat. Commun. 2018, 9, 3368. [Google Scholar] [CrossRef]
- Dimitroff, C.J. I-branched carbohydrates as emerging effectors of malignant progression. Proc. Natl. Acad. Sci. USA 2019, 116, 13729–13737. [Google Scholar] [CrossRef]
- Verkerke, H.; Dias-Baruffi, M.; Cummings, R.D.; Arthur, C.M.; Stowell, S.R. Galectins: An ancient family of carbohydrate binding proteins with modern functions. Galectins Methods Protoc. 2022, 2442, 1–40. [Google Scholar]
- Liu, F.-T.; Stowell, S.R. The role of galectins in immunity and infection. Nat. Rev. Immunol. 2023, 23, 479–494. [Google Scholar] [CrossRef]
- Braeuer, R.R.; Shoshan, E.; Kamiya, T.; Bar-Eli, M. The sweet and bitter sides of galectins in melanoma progression. Pigment. Cell Melanoma Res. 2012, 25, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Ebrahim, A.H.; Alalawi, Z.; Mirandola, L.; Rakhshanda, R.; Dahlbeck, S.; Nguyen, D.; Jenkins, M.; Grizzi, F.; Cobos, E.; Figueroa, J.A. Galectins in cancer: Carcinogenesis, diagnosis and therapy. Ann. Transl. Med. 2014, 2, 88. [Google Scholar]
- Chakraborty, A.; Perez, M.; Carroll, J.D.; Antonopoulos, A.; Dell, A.; Ortega, L.; Mohammed, N.B.; Wells, M.; Staudinger, C.; Griswold, A. Hypoxia Controls the Glycome Signature and Galectin-8–Ligand Axis to Promote Protumorigenic Properties of Metastatic Melanoma. J. Investig. Dermatol. 2023, 143, 456–469.e8. [Google Scholar] [CrossRef]
- Hayashi, Y.; Jia, W.; Kidoya, H.; Muramatsu, F.; Tsukada, Y.; Takakura, N. Galectin-3 inhibits cancer metastasis by negatively regulating integrin β3 expression. Am. J. Pathol. 2019, 189, 900–910. [Google Scholar] [CrossRef]
- Radosavljevic, G.; Volarevic, V.; Jovanovic, I.; Milovanovic, M.; Pejnovic, N.; Arsenijevic, N.; Hsu, D.K.; Lukic, M.L. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol. Res. 2012, 52, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Boscher, C.; Dennis, J.W.; Nabi, I.R. Glycosylation, galectins and cellular signaling. Curr. Opin. Cell Biol. 2011, 23, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Dennis, J.W. N-Glycans in cancer progression. Glycobiology 2008, 18, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Kariya, Y.; Kawamura, C.; Tabei, T.; Gu, J. Bisecting GlcNAc residues on laminin-332 down-regulate galectin-3-dependent keratinocyte motility. J. Biol. Chem. 2010, 285, 3330–3340. [Google Scholar] [CrossRef]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, e20182041. [Google Scholar] [CrossRef]
- Dange, M.C.; Bhonsle, H.S.; Godbole, R.K.; More, S.K.; Bane, S.M.; Kulkarni, M.J.; Kalraiya, R.D. Mass spectrometry based identification of galectin-3 interacting proteins potentially involved in lung melanoma metastasis. Mol. Biosyst. 2017, 13, 2303–2309. [Google Scholar] [CrossRef]
- Zhuo, Y.; Chammas, R.; Bellis, S.L. Sialylation of β1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J. Biol. Chem. 2008, 283, 22177–22185. [Google Scholar] [CrossRef]
- Ritsma, L.; Dey-Guha, I.; Talele, N.; Sole, X.; Salony; Chowdhury, J.; Ross, K.N.; Ramaswamy, S. Integrin β1 activation induces an anti-melanoma host response. PLoS ONE 2017, 12, e0175300. [Google Scholar] [CrossRef]
- Wellbrock, C.; Arozarena, I. The complexity of the ERK/MAP-kinase pathway and the treatment of melanoma skin cancer. Front. Cell Dev. Biol. 2016, 4, 33. [Google Scholar] [CrossRef]
- Barkan, D.; Chambers, A.F. β1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef]
- Guo, W.; Wang, H.; Li, C. Signal pathways of melanoma and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 424. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.; Chakraborty, A.; Lau, L.; Mohammed, N.; Dimitroff, C. Melanoma-associated glycosyltransferase GCNT2 as an emerging biomarker and therapeutic target. Br. J. Dermatol. 2021, 185, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.I.; Stegmayr, J.; Grant, O.C.; Yang, Z.; Nilsson, U.J.; Boos, I.; Carlsson, M.C.; Woods, R.J.; Unverzagt, C.; Leffler, H. Galectin binding to cells and glycoproteins with genetically modified glycosylation reveals galectin–glycan specificities in a natural context. J. Biol. Chem. 2018, 293, 20249–20262. [Google Scholar] [CrossRef] [PubMed]
- Mkhikian Haik, S.M.; Dennis James, W. The Galectin-lattice: A Decoder of bio-equivalent Glycans. Glycoforum 2021, 24, A10. [Google Scholar]
- Patnaik, S.K.; Potvin, B.; Carlsson, S.; Sturm, D.; Leffler, H.; Stanley, P. Complex N-glycans are the major ligands for galectin-1,-3, and-8 on Chinese hamster ovary cells. Glycobiology 2006, 16, 305–317. [Google Scholar] [CrossRef]
- Lagana, A.; Goetz, J.G.; Cheung, P.; Raz, A.; Dennis, J.W.; Nabi, I.R. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumor cells. Mol. Cell. Biol. 2006, 26, 3181–3193. [Google Scholar] [CrossRef]
- Kadirvelraj, R.; Yang, J.-Y.; Kim, H.W.; Sanders, J.H.; Moremen, K.W.; Wood, Z.A. Comparison of human poly-N-acetyl-lactosamine synthase structure with GT-A fold glycosyltransferases supports a modular assembly of catalytic subsites. J. Biol. Chem. 2021, 296, 100110. [Google Scholar] [CrossRef] [PubMed]
- Link-Lenczowski, P.; Lityńska, A. Glycans in melanoma screening. Part 2. Towards the understanding of integrin N-glycosylation in melanoma. Biochem. Soc. Trans. 2011, 39, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.C.F.; Andrade, L.N.D.S.; Bustos, S.O.; Chammas, R. Galectin-3 determines tumor cell adaptive strategies in stressed tumor microenvironments. Front. Oncol. 2016, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Stawikowski, M.J.; Aukszi, B.; Stawikowska, R.; Cudic, M.; Fields, G.B. Glycosylation modulates melanoma cell α2β1 and α3β1 integrin interactions with type IV collagen. J. Biol. Chem. 2014, 289, 21591–21604. [Google Scholar] [CrossRef]
- Mohammed, N.B.; Antonopoulos, A.; Dell, A.; Haslam, S.M.; Dimitroff, C.J. The pleiotropic role of galectin-3 in melanoma progression: Unraveling the enigma. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2023; Volume 157, pp. 157–193. [Google Scholar]
- Mohammed, N.B.; Lau, L.S.; Souchak, J.; Qiu, S.; Ahluwalia, M.S.; Osman, I.; Dimitroff, C.J. Tumor-Intrinsic Galectin-3 Suppresses Melanoma Metastasis. J. Investig. Dermatol. 2024, 144, 2039–2051.e9. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Mahi, N.A.; Najafabadi, M.F.; Pilarczyk, M.; Kouril, M.; Medvedovic, M. GREIN: An interactive web platform for re-analyzing GEO RNA-seq data. Sci. Rep. 2019, 9, 7580. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohammed, N.B.B.; Shil, R.K.; Dimitroff, C.J. Melanoma Glycome Regulates the Pro-Oncogenic Properties of Extracellular Galectin-3. Int. J. Mol. Sci. 2025, 26, 4882. https://doi.org/10.3390/ijms26104882
Mohammed NBB, Shil RK, Dimitroff CJ. Melanoma Glycome Regulates the Pro-Oncogenic Properties of Extracellular Galectin-3. International Journal of Molecular Sciences. 2025; 26(10):4882. https://doi.org/10.3390/ijms26104882
Chicago/Turabian StyleMohammed, Norhan B. B., Rajib K. Shil, and Charles J. Dimitroff. 2025. "Melanoma Glycome Regulates the Pro-Oncogenic Properties of Extracellular Galectin-3" International Journal of Molecular Sciences 26, no. 10: 4882. https://doi.org/10.3390/ijms26104882
APA StyleMohammed, N. B. B., Shil, R. K., & Dimitroff, C. J. (2025). Melanoma Glycome Regulates the Pro-Oncogenic Properties of Extracellular Galectin-3. International Journal of Molecular Sciences, 26(10), 4882. https://doi.org/10.3390/ijms26104882